Hodgkin-Lymphom











Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
Loslegennach dem britischen Pathologen Thomas Hodgkin (1798 bis 1866)
Synonyme: Morbus Hodgkin (obsolet), Lymphogranulomatose (obsolet)
Englisch: Hodgkin lymphoma, Hodgkin's disease
Definition
Beim Hodgkin-Lymphom handelt es sich um eine maligne lymphatische Systemerkrankung.
Geschichte
Der Pathologe Thomas Hodgkin präsentierte 1832 in London erstmalig den klinischen Verlauf sowie die Obduktionsbefunde von mehreren Patienten mit großen Lymphomen und Splenomegalie. Die pathologisch veränderten Lymphknoten waren entlang der großen Gefäße an Hals, Thorax und Abdomen zu finden. Später wurden charakteristische mehrkernige Riesenzellen gefunden, die heute als Sternberg-Reed-Zellen bezeichnet werden.
Epidemiologie
Die Krankheitsinzidenz in Deutschland beträgt etwa 3 Personen pro 100.000 Einwohner. Das Hodgkin-Lymphom stellt mit ca. 10 % eine der häufigsten Krebserkrankungen beim jungen Erwachsenen dar. Ein erster Altersgipfel liegt um das 32. Lebensjahr, ein zweiter tritt später um das 60. Lebensjahr auf.
Weiterhin zählt das Hodgkin-Lymphom zu den häufigsten nicht AIDS-definierenden Malignomen. Die Inzidenz ist bei HIV-Patienten um den Faktor 10 erhöht.
Ätiologie
Das Hodgkin-Lymphom ist eine maligne Erkrankung des lymphatischen Systems, wobei sich die neoplastischen Zellen fast immer von B-Lymphozyten ableiten.
Die genaue Ursache ist aktuell (2026) nicht vollständig geklärt. Eine Assoziation mit dem Epstein-Barr-Virus (EBV) wird vermutet, da entsprechende DNA in rund 50 %, bei HIV-Patienten sogar in über 90 % d.F. in den Tumorzellklonen nachgewiesen werden kann. Eine genetische Prädisposition wird vermutet, da familiäre Häufungen und ethnische Einflüsse beschrieben sind. Außerdem zählen Rauchen und ein hoher sozioökonomischer Status zu den bekannten Risikofaktoren.
Symptome
Patienten mit einem Hodgkin-Lymphom präsentieren sich in 70 % der Fälle mit einer schmerzlosen, derben bzw. gummiartigen Lymphknotenschwellung. Dabei können folgende Lokalisationen betroffen sein:
- zervikal (70 %), axillär (30 %), inguinal (10 %): sichtbare Schwellung
- mediastinal (60 %): ggf. Reizhusten, retrosternaler Druckschmerz, Dyspnoe, obere Einflussstauung
- abdominal (seltener): ggf. Oberbauchschmerzen, Cholestase oder Harnstau
Bei 40 % der Patienten treten B-Symptome auf. Dazu zählen:
- nicht anderweitig erklärbares Fieber über 38 °C: z.B. als undulierendes Pel-Ebstein-Fieber
- nicht anderweitig erklärbarer Nachtschweiß (mit Wechsel der Nachtwäsche)
- nicht anderweitig erklärbarer Gewichtsverlust von > 10 % des Körpergewichts in 6 Monaten
Weitere unspezifische Allgemeinsymptome sind Leistungsminderung und Pruritus. Charakteristisch, jedoch selten (5 %) sind Lymphknoten-Schmerzen nach Alkoholkonsum (Alkoholschmerz). Weitere Symptome finden sich bei ausgedehntem Befall bzw. Organbeteiligung:
- Veränderungen des Blutbildes bei Befall des Knochenmarks
- neurologische Symptome
- endokrine Störungen
- Knochenschmerzen
- Hepato- oder Splenomegalie
Zusätzlich können paraneoplastische Syndrome auftreten, z.B. eine autoimmunhämolytische Anämie, ein nephrotisches Syndrom oder ein Ophelia-Syndrom.
Diagnostik
Mittels detailierter Anamnese (v.a. Erfragen der B-Symptomatik) und einer körperlichen Untersuchung (u.a. Palpation der Lymphknoten und des Abdomens sowie Inspektion des Waldeyer-Rachenrings) ergeben sich Hinweise auf ein Hodgkin-Lymphom.
Zu den häufigen Laborbefunden zählen z.B.:
- BSG-, CRP- und LDH-Erhöhung
- Lymphozytopenie (< 1.000/μl)
- Eosinophilie (15 % d.F.)
Diese Veränderungen sind jedoch uncharakteristisch und erlauben keine Diagnose.
Die Diagnosesicherung erfolgt durch eine histologische Untersuchung eines Lymphknotens oder eines anderen primär befallenen Organs. Wenn möglich, sollte der Lymphknoten dabei im Ganzen entnommen werden. Eine Feinnadelaspiration mit anschließender zytologischer Untersuchung ist nicht ausreichend. Grundsätzlich sollte jede unklare Lymphknotenschwellung, die länger als vier Wochen persistiert oder progredient verläuft durch eine Lymphknoten-Exstirpation und histologische Untersuchung abgeklärt werden.
Anschließend erfolgt eine weitere Diagnostik zur Stadienerhebung (Staging), anhand derer die Therapie festgelegt wird.
Histopathologie
Die histopathologische Einteilung des Hodgkin-Lymphoms erfolgt nach der WHO-Klassifikation.
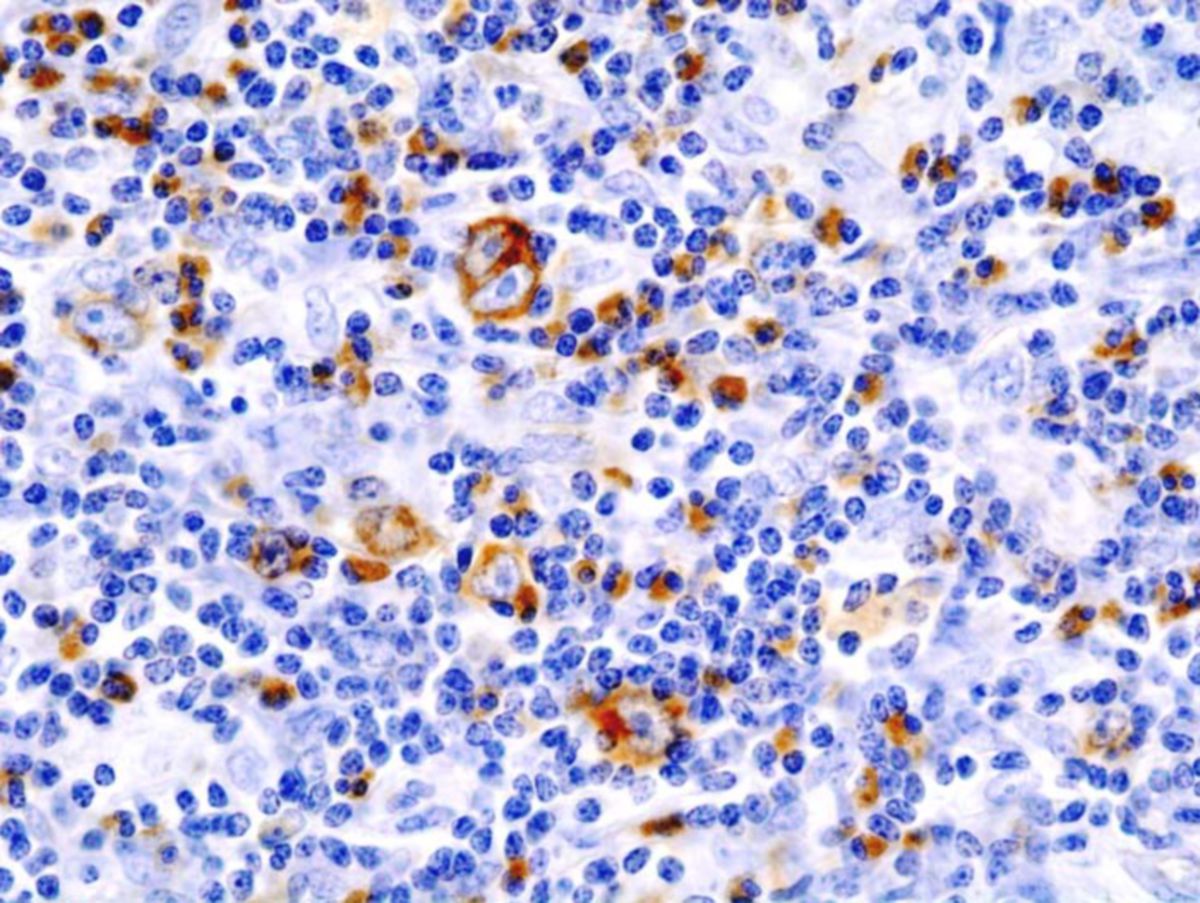
Klassisches Hodgkin-Lymphom
Das klassische Hodgkin-Lymphom (cHL) ist mit 95 % d.F. der häufigste Subtyp. Es zeichnet sich durch einkernige Hodgkin-Zellen und mehrkernige Sternberg-Reed-Zellen aus, die zusammenfassend als Hodgkin-Reed-Sternberg-Zellen (HRS-Zellen) bezeichnet werden. Diese Tumorzellen tragen typischerweise die Antigene CD30 und CD15 und machen lediglich 1 % der Gesamtzellmasse aus. Bei den tumorumgebenden Zellen (Bystander-Zellen) handelt es sich v.a. um CD3+- und CD4+-T-Lymphozyten.
Das cHL wird weiter in verschiedene Typen eingeteilt, wobei diese Subklassifikation keine therapeutische Relevanz besitzt.
Nodulär-sklerosierender Typ
Der nodulär-sklerosierende Typ (NS, noduläre Sklerose) stellt den häufigsten Subtyp dar (65 % d.F.). Es zeichnet sich durch Lakunarzellen (Sonderform der HRS-Zellen) sowie Kollagenfasern aus, die das lymphathische Gewebe durchsetzen und es in ein nodulär (knotig) geprägtes Muster unterteilen.
Klinisch befällt der nodulär-sklerosierende Typ bevorzugt Lymphknoten am unteren Hals, supraklavikulär und im Mediastinum.[1] Betroffen sind junge Erwachsene und Frauen etwas häufiger als Männer.
Mischtyp
Der Mischtyp (MC) ist insgesamt der zweithäufigste Typ (25 %), bei über 50 Jahre alten Patienten sowie bei HIV-Patienten sogar die häufigste Form des Hodgkin-Lymphoms. Männer sind häufiger betroffen als Frauen. Der Mischtyp befällt bevorzugt zervikale und abdominale Lymphknotenregionen.[1]
Histologisch zeigt sich ein gemischtzelliges Infiltrat aus Lymphozyten, Histiozyten, Granulozyten und HRS-Zellen sowie eine fein-fibrilläre Fibrose. Im Vergleich zu den anderen Formen des Hodgkin-Lymphoms ist der Mischtyp bei Diagnosestellung häufiger in einem weiter fortgeschrittenen Stadium. Die Patienten zeigen die prognostisch ungünstige B-Symptomatik entsprechend häufiger.
Lymphozytenreicher Typ
Der lymphozytenreiche Typ (LR) ist eher selten (4 %) und manifestiert sich meistens als Befall peripherer Lymphknoten.[1] Er kommt gehäuft bei männlichen Patienten um das 30. Lebensjahr vor und hat eine sehr gute Prognose.
Histologisch zeigt sich ein lymphozytenreiches Infiltrat (v.a. T-Lymphozyten) in Marginal- und Mantelzonen der Lymphfollikel sowie eine diffuse Fibrose.
Lymphozytenarmer Typ
Der lymphozytenarme Typ (LD) ist sehr selten (< 1 %) und manifestiert sich meistens durch abdominalen Lymphknotenbefall[1] und hat die schlechteste Prognose aller Typen des M. Hodgkin. Er ist fast immer mit EBV assoziiert und betrifft v.a. Frauen im Alter von 70 bis 80 Jahren.
Histologisch zeigt sich ein Bild von z.T. atypischen Hodgkin-Reed-Sternbergzellen und relativ wenigen Lymphozyten.
Noduläres lymphozytenprädominantes Hodgkin-Lymphom
Das noduläre lymphozytenprädominante Hodgkin-Lymphom (NLPHL, LPHD) macht ca. 5 % der Hodgkin-Lymphome aus. Die Tumorzellen werden auch Popcorn-Zellen genannt. Sie stellen eine Variante der HRS-Zelle mit gelapptem Zellkern dar, die meist die B-Zell-Antigene CD20 und CD79a tragen.
Staging
Die diagnostischen Untersuchungen umfassen folgende Aspekte, wobei das Staging innerhalb von vier Wochen nach histologischer Diagnosesicherung abgeschlossen sein sollte:
- Anamnese: B-Symptomatik, weitere Symptome, Allgemeinzustand (Aktivitätsindex nach WHO), Begleiterkrankungen, Familienanamnese
- körperliche Untersuchung: Palpation der Lymphknoten, Milz, Leber und des Abdominalbereichs
- Labor:
- Blutbild mit Differenzialblutbild
- BSG
- GOT, GPT, gGT, AP, LDH, Bilirubin, Harnsäure, Kreatinin
- HIV-Antikörpersuchtest, Hepatitis-B- und -C-Diagnostik
- β-HCG bei Frauen
- Bildgebung:
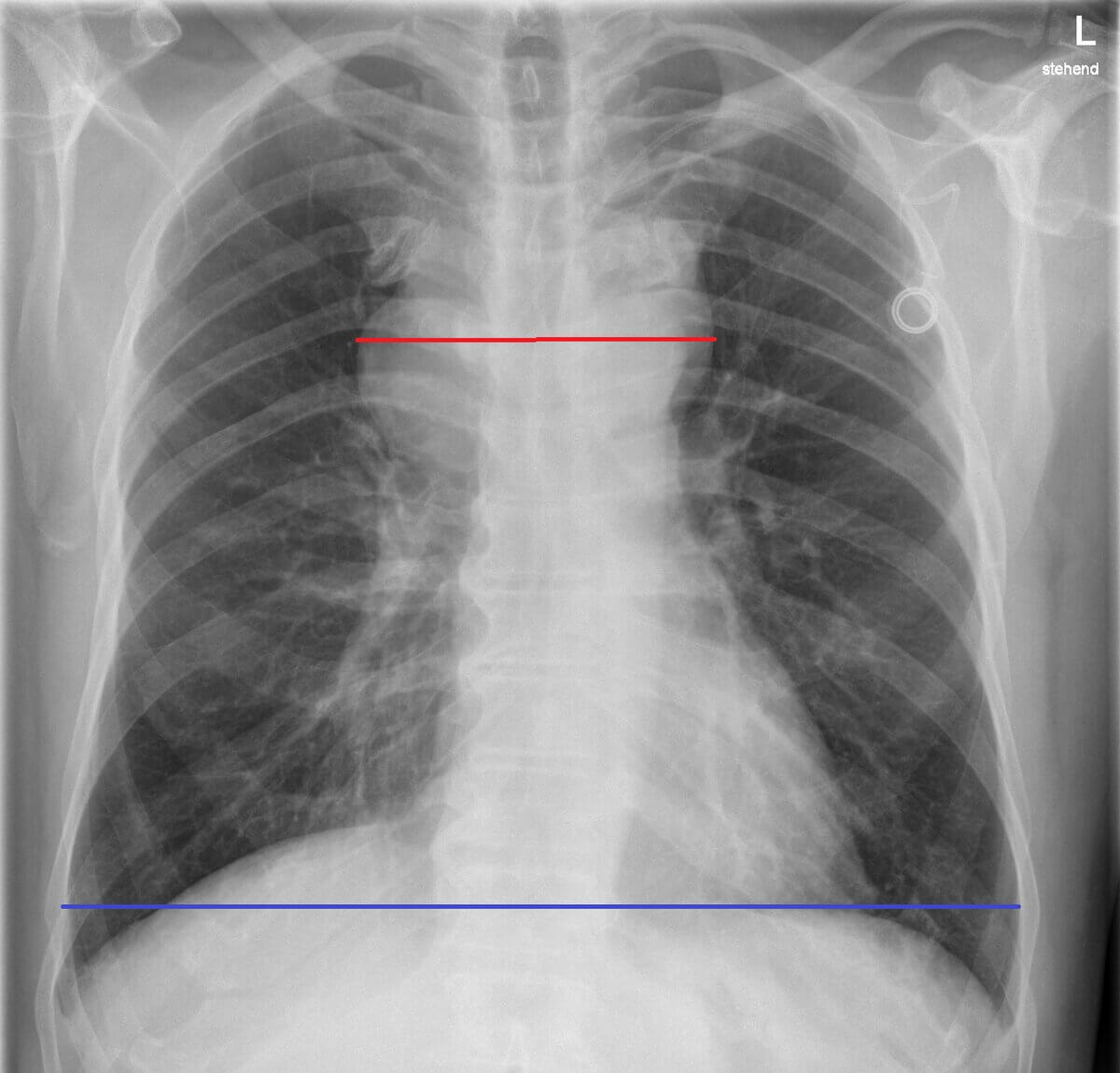
- Röntgen-Thorax: p.a.-Aufnahme zum Ausschluss eines großen Mediastinaltumors
- CT-Hals, -Thorax und -Abdomen mit Kontrastmittel: verdächtig sind Lymphknoten > 10 mm bzw. inguinale Lymphknoten > 15 mm, insbesondere wenn diese kugelig, ohne Fetthilus, mit zentraler Nekrose und/oder vermehrt vorliegen; bei Kontrastmittel-Unverträglichkeit alternative Bildgebung (z.B. MRT)
- Ganzkörper-PET: falls verfügbar; im Rahmen der CT-Untersuchung (PET-CT)
- fakultativ: Sonographie des Halses und Abdomens
- Knochenmarkbiopsie: nur bei unklarem PET-CT-Befund
- Abklärung von verdächtigen extranodalen Läsionen (z.B. mittels Sonographie, MRT, CT oder Biopsie)
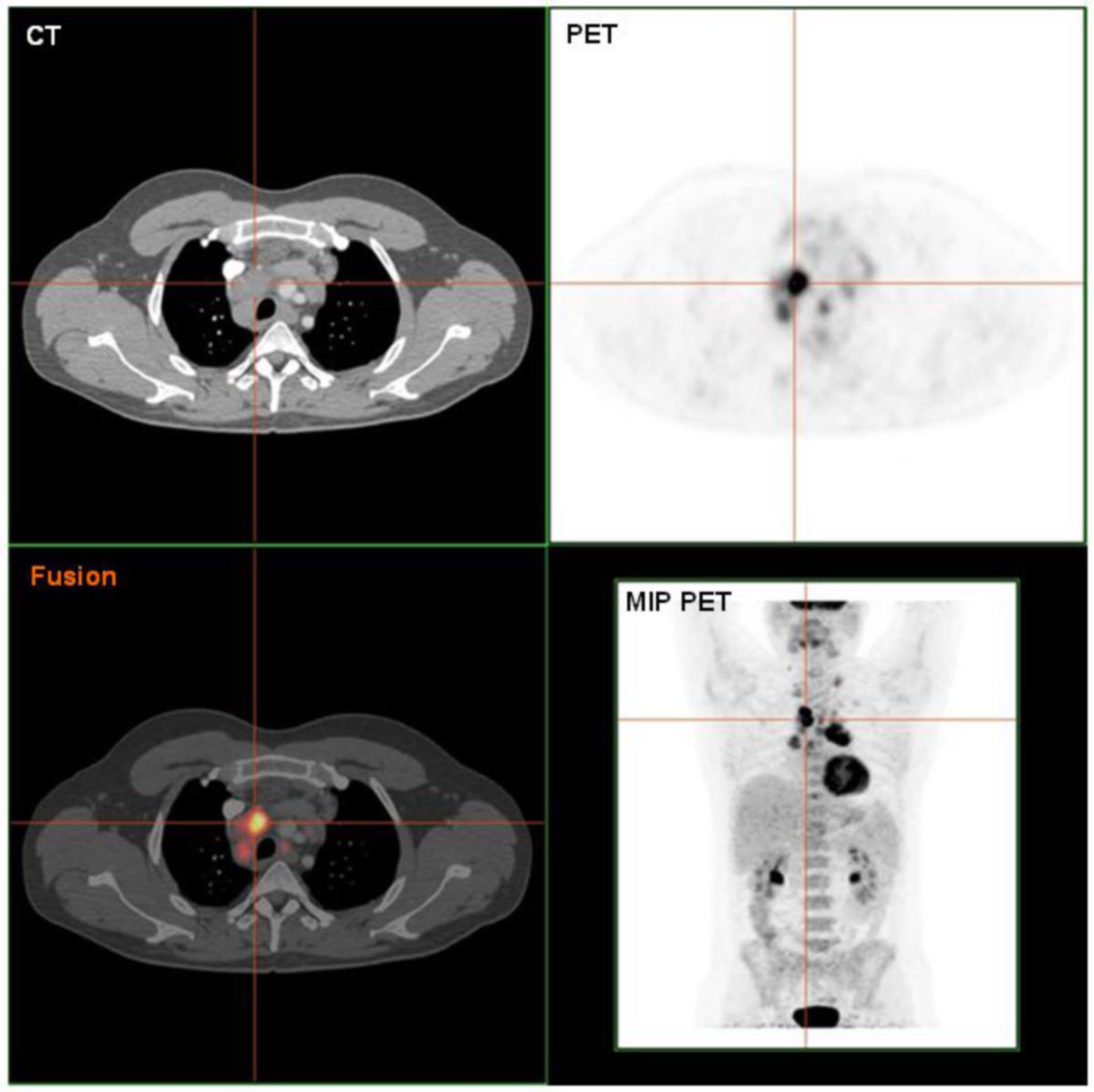
CT-Fallbeispiel
Prätherapeutische Toxizitätsdiagnostik
Zur Identifikation von Patienten mit einem erhöhten Risiko für Akut- und/oder Spätkomplikationen der Therapie ("Toxizitätsbeurteilung") sind weitere Untersuchungen notwendig. Dazu zählen:
- Lungenfunktionsdiagnostik
- EKG und Echokardiographie
- Schilddrüsenfunktionsdiagnostik: TSH-Bestimmung
- Fertilitätsdiagnostik:
- Bestimmung von FSH und LH
- bei Frauen: Dokumentation der Zyklusanamnese und Bestimmung von Anti-Müller-Hormon
- bei Männern: Spermiogramm bei nicht abgeschlossener Familienplanung, Bestimmung von Testosteron und Inhibin B
Bei nicht abgeschlossener Familienplanung sollte eine Beratung bzgl. fertilitätserhaltenden Maßnahmen angeboten werden. Bei pulmonaler Vorbelastung sollte auf Bleomycin, bei kardialen Vorerkrankungen auf Anthracycline verzichtet werden.
Stadieneinteilung
Die Stadieneinteilung des Hodgkin-Lymphoms erfolgt nach der Cotswold-modifizierten Ann-Arbor-Klassifikation:
| Stadium | Beschreibung |
|---|---|
| Stadium I | Lokaler Befall:
|
| Stadium II | Eine Seite des Zwerchfells befallen:
|
| Stadium III | Beide Seiten des Zwerchfells befallen:
|
| Stadium IV | Disseminierter Befall
|
| Zusatz A | keine B-Symptome |
| Zusatz B | mind. ein B-Symptom |
| lymphatisches Gewebe: Lymphknoten, Milz, Thymus, Waldeyer-Rachenring, Blinddarm, Peyer-Plaques | |
Risikostratifizierung
Die Deutsche Hodgkin Studiengruppe (GHSG) definiert weiterhin vier Risikofaktoren, anhand derer eine weitere Risikostratifizierung erfolgt:
- Befall von mindestens 3 Lymphknotenarealen (Achtung: Die Lymphknotenareale unterscheiden sich von den Lymphknotenregionen der Ann-Arbor-Klassifikation)
- hohe BSG (> 50 mm/h ohne B-Symptomatik, > 30 mm/h mit B-Symptomatik)
- großer Mediastinaltumor (mind. 1/3 des maximalen Thoraxquerdurchmessers im p.a.-Röntgen-Thorax)
- extranodaler Befall (außerhalb des lymphatischen Gewebes)
| IA, IB, IIA | IIB | III, IV | |
|---|---|---|---|
| kein Risikofaktor | frühes Stadium | fortgeschrittenes Stadium | |
| mind. 3 Lymphknotenareale und/oder hohe BSG | intermediäres Stadium | fortgeschrittenes Stadium | |
| Mediastinaltumor und/oder extranodaler Befall | intermediäres Stadium | fortgeschrittenes Stadium | |
Differenzialdiagnosen
Als Differenzialdiagnose müssen andere Erkrankungen erwogen werden, die mit einer Lymphknotenvergrößerungen einhergehen können. Dazu zählen z.B.:
- infektiöse Erkrankungen: z.B. CMV-, EBV-, HIV-Infektion, Tuberkulose, Listeriose, Leptospirose, Toxoplasmose
- nicht-infektiöse Erkrankungen: z.B. Metastasen, Non-Hodgkin-Lymphome, Sarkoidose, nekrotisierende Lymphadenitis
Weiterhin müssen auch Erkrankungen bedacht werden, die zu B-Symptomen führen können, beispielsweise Leukämien, Thymome oder eine rheumatoide Arthritis sowie ein systemischer Lupus erythematodes.
Bei einem mediastinalen Tumor muss auch an Keimzelltumoren, Schilddrüsenkarzinome, Neurinome oder an einen Morbus Castleman gedacht werden.
Therapie
Die Therapie erfolgt in spezialisierten Zentren möglichst im Rahmen von klinischen Studien. Je nach Stadieneinteilung und Risikofaktoren kommt eine Chemotherapie häufig kombiniert mit einer Strahlentherapie zum Einsatz. Typischerweise werden folgende Schemata eingesetzt:
- ABVD-Schema: Adriamycin (Doxorubicin), Bleomycin, Vinblastin, Dacarbazin
- BEACOPP-eskaliert-Schema: Bleomycin, Etoposid, Adriamycin, Cyclophosphamid, Oncovin® (Vincristin), Procarbazin, Prednisolon
- BrECADD-Schema: Brentuximab-Vedotin, Etoposid, Cyclophosphamid, Adriamycin, Dacarbazin, Dexamethason
Die Bestrahlung erfolgt heutzutage (2026) in Form einer lokalen Involved-Site-Radiatio (ISRT). In Ausnahmefällen kann eine Protonenbestrahlung erwogen werden.
Kurative Therapie
Die Therapie des Hodgkin-Lymphoms erfolgt stets kurativ, unabhängig vom Stadium.
| Stadium | Chemotherapie | Adjuvante Bestrahlung |
|---|---|---|
| Frühes Stadium | 2 Zyklen ABVD | 20 Gy IS-RT von prätherapeutisch befallenen Lokalisationen |
| Intermediäres Stadium |
|
30 Gy IS-RT von prätherapeutisch befallenen Lokalisationen |
| Fortgeschrittenes Stadium |
|
30 Gy IS-RT von PET-positiven Resten (> 1,5 cm) |
Pallative Therapie
Komorbide Patienten, die für eine Polychemotherapie nicht in Frage kommen, werden i.d.R. palliativ behandelt. Eingesetzt werden Gemcitabin, Vinorelbin sowie eine lokale Strahlentherapie.
Rezidivtherapie
Die meisten Patienten mit einem histologisch gesicherten Rezidiv werden mittels Hochdosischemotherapie (z.B. BEAM-Schema: Carmustin, Etoposid, Cytarabin, Melphalan) und anschließender autologer Stammzelltransplantation (SZT) behandelt. Vor Hochdosistherapie sollte eine Salvage-Therapie bzw. Reinduktionstherapie erfolgen. Diese wird i.d.R. mit zwei Zyklen DHAP (Dexamethason, Hochdosis-Cytarabin, Cisplatin) durchgeführt. Anschließend erfolgt eine PET-CT-Untersuchung: Bei Remission oder stabiler Erkrankung sollte zeitnah die Therapie fortgeführt werden. Bei Progress kann eine alternative Salvage-Therapie oder Brentuximab-Vedotin eingesetzt werden.
Bei Patienten über 60 Jahre wird i.d.R. eine konventionelle Chemotherapie und/oder eine lokale Bestrahlung empfohlen. Bei Hochrisikopatienten (z.B. Stadium IV bei Rezidiv) ist beispielsweise eine Erhaltungstherapie mit Brentuximab-Vedotin möglich.
Weiterhin existieren folgende Therapieoptionen:
- alleinige Strahlentherapie: bei lokalisiertem Spätrezidiv (> 12 Monate nach erfolgreicher Primärtherapie) außerhalb des initialen Strahlenfeldes sowie ohne B-Symptome oder Anämie
- Rezidivtherapie mit erneuter Erstlinientherapie: bei Spätrezidiv nach Erstdiagnose initial im frühen Stadium
- erneute Stammzelltransplantation: bei Spätrezidiv nach autologer Stammzelltransplantation
- Brentuximab-Vedotin: bei refraktärem oder rezidivierendem CD30+-Hodgkin-Lymphom nach Versagen der autologen Stammzelltransplantation bzw. zweier vorangegangener Therapien
- PD-1-Inhibitoren (Nivolumab, Pembrolizumab): bei refraktärem oder rezidivierendem Hodgkin-Lymphom nach Versagen einer autologen Stammzelltransplantation
- allogene Stammzelltransplantation: bei Rezidiv nach autologer Transplantation und zumindest partieller Remission bei Patienten in gutem Allgemeinzustand
Therapie bei NLPHL
Patienten mit NLPHL im Stadium IA ohne Risikofaktoren besitzen eine sehr gute Prognose. In diesem Fall ist eine alleinige Bestrahlung mit 30 Gy ausreichend. Alle anderen NLPHL-Patienten werden analog zum cHL behandelt.
Bei Patienten mit einem Rezidiv eines NLPHL sollte eine erneute Diagnosesicherung mittels Lymphknotenbiopsie erfolgen, da ein Risiko für die Transformation in ein aggressives Non-Hodgkin-Lymphom besteht. Analog zu den Patienten mit cHL kann eine Hochdosischemotherapie mit nachfolgender autologer Stammzelltransplatation oder off label ein Anti-CD20-Antikörper (z.B. Rituximab) erwogen werden.
Nachsorge
Asymptomatische Patienten erhalten eine Nachsorgeuntersuchung alle 3 Monate, im zweiten bis vierten Jahr alle 6 Monate und ab dem fünften Jahr einmal jährlich. Obligate Maßnahmen sind:
- Anamnese
- körperliche Untersuchung
- Labor: Differenzialblutbild, BSG, klinische Chemie
- CT-Untersuchung nur bei partieller Remission 3 Monate nach Abschluss-Staging sowie bei klinischem Verdacht auf ein Rezidiv
Fakultativ kann eine Sonographie eingesetzt werden. Bei Verdacht auf ein Rezidiv in der Bildgebung sollte eine histologische Sicherung angestrebt werden.
Die Nachsorge dient nicht nur der Entdeckung eines Rezidivs, sondern auch zur Kontrolle von ggf. auftretenden Spätkomplikationen der Chemo- und Strahlentherapie:
- Kardiotoxizität: Kardiale Erkrankungen gehören zu den häufigsten Todesursachen bei Langzeitüberlebendes eines Hodgkin-Kymphoms. Ursächlich sind neben einer mediastinalen Bestrahlung die Behandlung mit Anthracyclinen. Daher ist eine gezielte kardiale Nachsorge notwendig.
- Schilddrüsenstörungen: Eine Hypothyreose ist eine häufige Spätfolge nach lokaler Bestrahlung (Inzidenz 35 %), weiterhin besteht ein erhöhtes Risiko für die Entwicklung eines Morbus Basedow, insbesondere bei Strahlentherapie, daher einmal jährliche Bestimmung von TSH
- Pulmonale Störungen: Risiko einer Lungenfibrose bei Bleomycin oder mediastinaler Bestrahlung, daher 12 Monate nach Therapieende Lungenfunktionsprüfung (mit Röntgen-Thorax und Ermittlung der Diffusionskapazität bei entsprechenden Symptomen)
- Gonadale Toxizität: Durch die Strahlen- und Chemotherapie kann es zur Beeinträchtigung der Fortpflanzungsfähigkeit kommen, insbesondere bei Strahlentherapie und Alkylantien
- Fatigue: Prävalenz von ca. 40 %, selbst Jahre nach Therapieabschluss; nach Ausschluss anderer organischer und psychiatrischer Ursachen erfolgt eine Therapie durch Sportinterventionen und psychoonkologische Maßnahmen
- Sekundärmalignome: abhängig von der kumulativen Dosis und der Auswahl der Zytostatika sowie der Dosis und Feldgröße der Strahlentherapie
- sekundäre akute myeloische Leukämie und myelodysplastisches Syndrom: Inzidenz 0,5 bis 2 %, dabei 25 % d.F. innerhalb des ersten Jahres, 80 % d.F. in den ersten 5 Jahren nach Behandlung, Gesamtüberleben nach 2 Jahren ca. 8 %
- Non-Hodgkin-Lymphome: treten mit einer Latenzzeit von 5 bis 15 Jahren auf
- Bronchialkarzinom: macht ca. 25 % der Sekundärneoplasien aus
- Mammakarzinom: häufigste Sekundärneoplasie bei Frauen, v.a. wenn diese bei Erstdiagnose unter 30 Jahre alt waren; in diesem Fall ab 8 Jahre nach Therapie regelmäßiges Screening (halbjährlich durch Palpation und Sonographie sowie jährlich mittels MR-Mammographie); individuelles Screening bei Frauen, die zum Diagnosezeitpunkt über 30 Jahre alt waren
- Kolorektales Karzinom: erhöhtes Risiko 10 bis 15 Jahre früher als in der Normalbevölkerung, bisher kein frühzeitiges Screening empfohlen
- Schilddrüsenkarzinome
Prognose
Die Prognose des Hodgkin-Lymphoms ist in allen Stadien bei adäquater Therapie sehr gut. Auch bei Patienten, bei denen die Krankheit bereits fortgeschritten ist, wird in rund 80 % der Fälle eine Remission erreicht. Die 5-Jahres-Überlebensrate liegt bei über 90 % und nach einem Rezidiv bei 50 %.
Somit gehört das Hodgkin-Lymphom zu den am besten behandelbaren onkologischen Erkrankungen im Erwachsenenalter.
Quiz
Literatur
- Onkopedia Leitlinie Hodgkin-Lymphom, Stand 01.2018, abgerufen am 12.11.2019
- DGHO, AWMF S3-Leitline, April 2019, abgerufen am 12.11.2019
Bidquellen
- Bildquelle DICOM-Viewer: Idris et al (2024). Mediastinal Lymph Node Quantification (LNQ): Segmentation of Heterogeneous CT Data (Version 1) case_0064. The Cancer Imaging Archive
- Bildquelle für Flexikon-Quiz: © Rupert Britton / [1]