Systemischer Lupus erythematodes











Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenSynonyme: Lupus erythematodes disseminatus, LED
Englisch: systemic lupus erythematosus
Definition
Beim systemischen Lupus erythematodes, kurz SLE, handelt es sich um eine Systemerkrankung, bei der Haut und Gefäßbindegewebe der Organe durch Vaskulitiden und Ablagerungen von Immunkomplexen betroffen sind. Der SLE zählt zur Gruppe der Kollagenosen.
siehe auch: Lupus erythematodes
Abgrenzung
Die systemische Form des Lupus erythematodes ist besonders durch die Organbeteiligung gekennzeichnet. Sie wird abgegrenzt von den verschiedenen Formen des kutanen Lupus erythematodes (CLE), die nur einen Befall der Haut aufweisen und daher eine bessere Prognose haben. Die Schwäche dieser Abgrenzung ist, dass sie rein deskriptiv auf dem klinischen Bild basiert, nicht auf "harten" pathogenetischen Parametern. Die Übergänge zwischen der kutanen und der systemischen Form sind fließend. So konnte im Verlauf von 8 Jahren bei 17 % der CLE-Patienten ein Übergang von der kutanen zur systemischen Form beobachtet werden.[1]
Eine weitere Form des Lupus erythematodes ist der medikamentös induzierte Lupus (DIL), der durch eine Vielzahl von Medikamenten ausgelöst werden kann. Die Symptomatik kann dem SLE sehr ähnlich sein. Ein Auftreten antinukleärer Antikörper (ANA) ist typisch. Im Gegensatz zum SLE finden sich hier jedoch keine dsDNS-Antikörper.
Epidemiologie
Ätiologie
Die Ursachen für den SLE sind bislang (2024) nicht geklärt. Man vermutet derzeit, dass die Erkrankung bei Menschen mit genetischer Prädisposition durch bestimmte Umweltfaktoren ausgelöst wird. Epigenetische Modifikationen vermitteln dabei die immunologische Reaktion auf diese Faktoren. Die Erblichkeit wird auf etwa 44 % geschätzt und beschreibt den Anteil von phänotypischen Varianzen, die durch genetische Faktoren erklärt werden können.
Ein erhöhtes Risiko besteht bei der Exposition mit Zigarettenrauch, oralen Kontrazeptiva und Hormonersatztherapien. Noch unzureichend geklärt ist der Einfluss von z.B. Pestiziden, Schwermetallen, UV-Licht, Infektionen und Übergewicht.
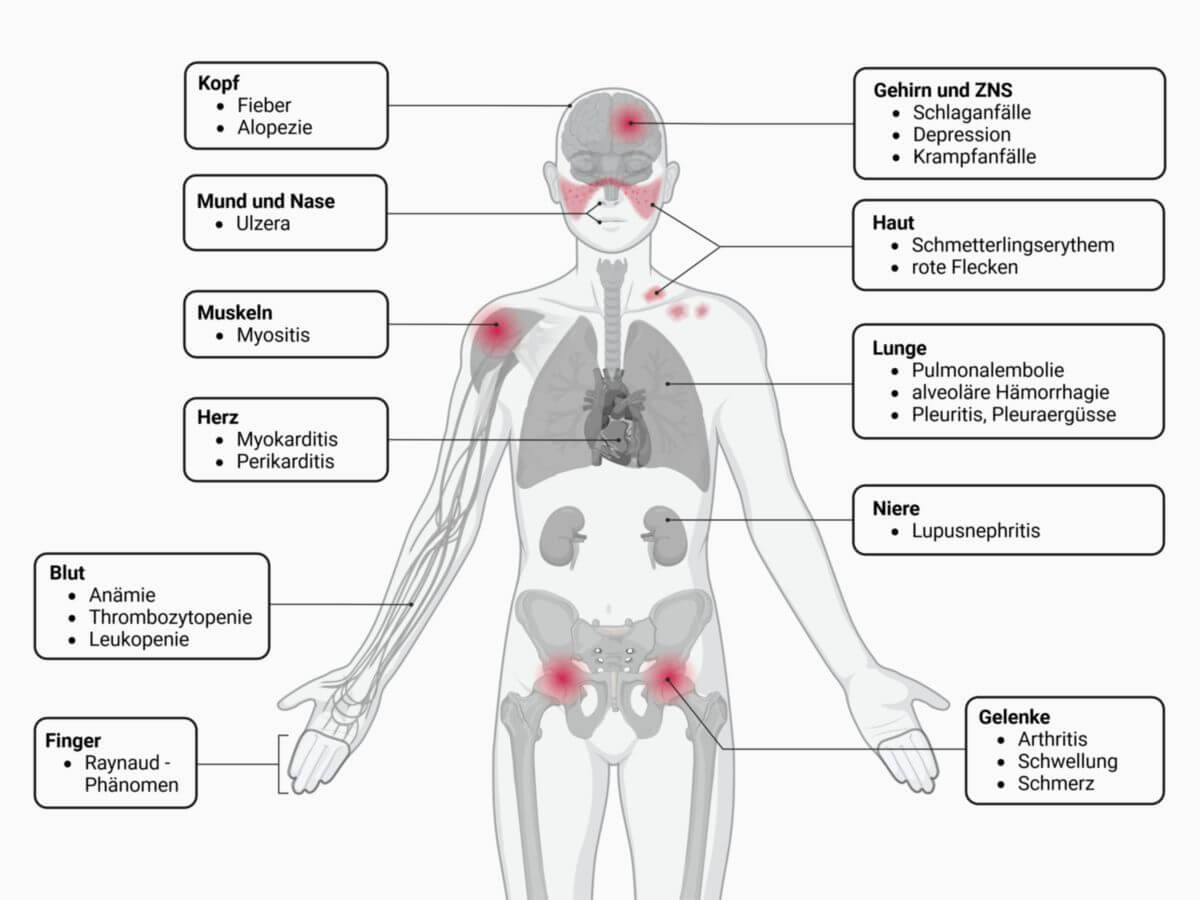
Symptome
Die Klinik des SLE ist interindividuell sehr variabel und kann verschiedene Organsysteme betreffen, wobei es zu unterschiedlichen Befallsmustern kommt. Die Erkrankung verläuft meist schubweise, permanent aktive Verläufe sind seltener. Die Symptome lassen sich in allgemeine und spezifische Symptome einteilen.
Allgemeinsymptome
Ungefähr 95 % aller Betroffenen leiden unter folgendem, allgemeinem Beschwerdebild:
- Fieber
- allgemeine Schwächezustände (ähnlich eines grippalen Infektes)
- Gewichtsabnahme
- Lymphadenopathie
Spezifische Symptome
- Bewegungsapparat
- Myositis (ca. 40 %)
- Polyarthritis (> 80 %)
- Haut
- Schmetterlingserythem: Auffallende Rötungen an Wangen und Nase
- Diskoider Lupus: Papeln mit Schuppenbildung
- vernarbende Alopezie
- Ulzera der oralen bzw. nasalen Schleimhäute
- Photosensibilität
- Lunge
- Pleuritis und Pleuraergüsse (30 %)
- interstitielle Lungenerkrankung (1 bis 15 %): meist NSIP-Muster, seltener UIP-, LIP- oder OP-Muster
- Lupuspneumonitis (1 bis 3 %): diffuse alveoläre Hämorrhagie mit Kapillaritis und diffusem Alveolarschaden
- Lungenarterienembolie
- pulmonale Hypertonie
- Herz: Myokarditis, Perikarditis, Libman-Sacks-Endokarditis
- Niere: Lupusnephritis (Immunkomplexnephritis)
- Nervensystem: Depression, Krampfanfälle, Schlaganfall
- Blut: autoimmunhämolytische Anämie, Thrombozytopenie, Leukopenie
- Finger: Raynaud-Phänomen
Diagnostik
Die Diagnose wird neben dem klinischen Erscheinungsbild vor allem durch die Labordiagnostik gestellt:
Entzündungszeichen
- erhöhte BSG
- Alpha 2/gamma Globuline erhöht
- Komplementfaktoren C3 und C4 erniedrigt
- hypochrome Anämie
- CRP normal
Immunologisches Labor
- Antinukleäre Antikörper (ANA)
- DsDNA-Antikörper: Autoantikörper gegen doppelsträngige DNA
- Anti-PCNA: Autoantikörper gegen Proliferating-Cell-Nuclear-Antigen
- Sm-Antikörper: Autoantikörper, die gegen Komponenten der snRNPs gerichtet sind
- Antiphospholipid-Antikörper: Der Nachweis von Anti-Cardiolipin-AK und Lupus-Antikoagulans weist auf APA hin. Bei dieser Konstellation kann das sogenannte Antiphospholipid-Syndrom auftreten.
Diagnosekriterien
Die Diagnose eines SLE wird anhand der ACR/EULAR-Klassifikationskriterien (2019) gestellt, gelegentlich auch anhand der älteren SLICC-Kriterien (2012). Die ACR/EULAR-Kriterien sind in der folgenden Tabelle aufgeführt. Die Diagnose kann bei einer Punktzahl von ≥ 10 Punkten gestellt werden. Dabei sind folgende Voraussetzungen und allgemeine Prinzipien zu beachten:
- ANA (HEp2-IFT) ≥ 1:80 (einmaliger Nachweis ist ausreichend)
- Ein Kriterium wird nicht gewertet, wenn eine andere, wahrscheinlichere Ursache zugrunde liegt (z.B. Infektion, Neoplasie, Medikamente oder andere Erkrankung)
- Ein Kriterium ist erfüllt, wenn es einmalig vorgekommen und dokumentiert ist
- Kriterien müssen nicht gleichzeitig vorliegen
- Es muss jedoch mindestens ein Kriterium aktuell vorhanden sein
- Innerhalb jeder Domäne geht nur der höchste Score in den Gesamtscore ein
| Klinische Domäne und Kriterien | Punkte | |
|---|---|---|
| Konstitutionelle Symptome | Fieber | 2 |
| Haut | nicht vernarbende Alopezie orale Ulzera subakut-kutaner (SCLE) oder diskoider LE (DLE) akuter kutaner LE (ACLE) |
2 2 4 6 |
| Arthritis | Synovitis in ≥ 2 Gelenken oder Druckschmerz in ≥ 2 Gelenken + Morgensteifigkeit ≥ 30 min. | 6 |
| Neurologie | Delirium Psychose Krampfanfälle |
2 3 5 |
| Serositis | Pleura- oder Perikarderguss Akute Perikarditis |
5 6 |
| Hämatologie | Leukopenie Thrombopenie Autoimmunhämolyse |
3 4 4 |
| Nieren | Proteinurie > 0,5 g/24h Lupusnephritis (histologisch) Typ II, V Lupusnephritis (histologisch) Typ III, IV |
4 8 10 |
| Immunologische Domänen und Kriterien: | ||
| Antiphospholipid-Antikörper | Anticardiolipin-Antikörper (aCL): > 40 GPL oder Beta-2-Glykoprotein I (aβ2-GPI): > 40 GPL oder Lupus-Antikoagulans (LA): + |
2 |
| Komplement | C3 ODER C4 vermindert C3 UND C4 vermindert |
3 4 |
| Hochspezifische Auto-Ak | anti-ds-DNA-AK oder anti-Sm-AK |
6 |
Krankheitsaktivität
Die Krankheitsaktivität des SLE kann mithilfe von Scoringsystemen erfasst werden. Ein Beispiel ist der Systemic Lupus Erythematosus Disease Activity Index (SLEDAI).
Therapie
Generell gilt es eine UV-Exposition zu vermeiden und auf einen adäquaten Lichtschutz zu achten. Je nach Stadium und Begleiterscheinung wird die Therapie des SLE darüber hinaus angepasst.
Leichte Form ohne viszeralen Befall
Die Therapie erfolgt mit NSAR und Hydroxychloroquin. Bei entzündlichen Schüben kommen zudem zeitlich begrenzt Kortikosteroide zum Einsatz. Alternativ können Immunsuppressiva eingesetzt werden.
Zudem ist der monoklonale Antikörper Belimumab als Begleitmedikation für den SLE mit Krankheitsaktivität unter Standardtherapie zugelassen. Bis zum Wirkeintritt zeigt sich allerdings eine gewisse Latenz von ≥ 3 Monaten. Belimumab ist somit nur bedingt für die Akuttherapie geeignet.
Schwere Form mit Organbeteiligung
Die Therapie erfolgt mit einer hochdosierten Prednisolon-Stoßtherapie und/oder Immunsuppressiva. In mittelschweren Fällen kommen MTX, Azathioprin oder Ciclosporin A, in schweren Fällen Cyclophosphamid zum Einsatz. Mycophenolat-Mofetil kann darüber hinaus bei Versagen der zuvor genannten Therapeutika oder bei entsprechenden Kontraindikationen indiziert sein. Darüber hinaus wird bei frustranen Verläufen auch Rituximab off-label als mögliche Therapie diskutiert. In therapierefraktären Fällen kommt darüber hinaus die Anwendung einer CAR-T-Zell-Therapie in Betracht, z.B. mit Rapcabtagen-Autoleucel (experimentelle Therapie).
Weiteres
Zur Vermeidung von Komorbiditäten wird die Behandlung z.B. mit Antihypertensiva, Lipidsenkern, Antikoagulanzien und knochenschützenden Medikamenten empfohlen.
Therapieziel
Ein konsensbasierter Zielwert für eine kontrollierte, niedrige Krankheitsaktivität ist der Lupus Low Disease Activity State (LLDAS).
Merkhilfe
Um sich das klinische Spektrum des SLE merken zu können, kann das Akronym SOAP-BRAIN-MD hilfreich sein.
- S: Serositis (Pleuritis, Perikarditis)
- O: Orale Ulzera
- A: Arthritis
- P: Photosensibilität
- B: Blut (Erniedrigung aller Parameter, d.h. Anämie, Leukopenie, Thrombozytopenie)
- R: Renal (Proteinurie)
- A: ANA
- I: Immunologische Störung (anti-ds-DNA-AK, anti-Sm-AK)
- N: Neurologische Störung (Krampfanfälle, Psychose, Delirium)
- M: Malar rash (Schmetterlingserythem)
- D: Discoid rash (Diskoides Erythem)
Literatur
- Kuhn A et al: Diagnostik und Therapie des systemischen Lupus Erythematodes (Übersichtsarbeit), Deutsches Ärzteblatt 2015, frei zugänglich
- Bertsias G, Cervera R, Boumpas DT: Systemic Lupus Erythematosus: Pathogenesis and Clinical Features
- Gergianaki I, Bertsias G: Systemic Lupus Erythematosus in Primary Care: An Update and Practical Messages for the General Practitioner. Front Med (Lausanne). 2018 May 29;5:161. doi: 10.3389/fmed.2018.00161.
- Kunz M: Lupus erythematosus. Part I: epidemiology, genetics and immunology. J Dtsch Dermatol Ges. 2013 Aug;11(8):709-19; quiz 720, 709-720; quiz 721. doi: 10.1111/ddg.12165.
- Felten R, Dervovic E, Chasset F, Gottenberg JE, Sibilia J, Scher F, Arnaud L.: The 2018 pipeline of targeted therapies under clinical development for Systemic Lupus Erythematosus: a systematic review of trials. Autoimmun Rev. 2018 Jun 6. pii: S1568-9972(18)30133-2. doi: 10.1016/j.autrev.2018.02.011
- Fanouriakis A et al. 2019 update of the EULAR recommendations for the management of systemic lupus erythematosus, Annals of the Rheumatic Diseases 2019;78:736-745, abgerufen am 18.10.2019
Quellen
- ↑ Wieczorek et al., Systemic Symptoms in the Progression of Cutaneous to Systemic Lupus Erythematosus, JAMA Dermatol. 2014