Lupus erythematodes









Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenEine neue Leitlinie ist verfügbar.
Klick auf "Bearbeiten" und aktualisiere diesen Artikel!
Eine neue Leitlinie ist verfügbar.
Klick auf "Bearbeiten" und aktualisiere diesen Artikel!
von lateinisch: lupus - Wolf; von altgriechisch: erythema - Röte
Synonym: Schmetterlingsflechte
Abkürzung: LE
Englisch: Lupus erythematosus
Definition
Lupus erythematodes, kurz LE, ist eine systemische Autoimmunerkrankung, die zusammen mit der systemischen Sklerose und der Dermatomyositis zu den Kollagenosen gehört. Typisch für diese Erkrankungen sind hohe Titer an Autoantikörpern, die sich gegen körpereigene Organe richten.
ICD-Klassifizierung
ICD-10-Codes:
- M32.-: Systemischer Lupus erythematodes
- M32.0: Arzneimittelinduzierter systemischer Lupus erythematodes
- M32.1+: Systemischer Lupus erythematodes mit Beteiligung von Organen oder Organsystemen
- M32.8: Sonstige Formen des systemischen Lupus erythematodes
- M32.9: Systemischer Lupus erythematodes, nicht näher bezeichnet
- L93.-: Lupus erythematodes
- L93.0: Diskoider Lupus erythematodes
- L93.1: Subakuter Lupus erythematodes cutaneus
- L93.2: Sonstiger lokalisierter Lupus erythematodes
Etymologie
Die häufige Beteiligung der Haut hat der Erkrankung ihren Namen gegeben: Lupus (= Wolf) steht in diesem Falle für den aggressiven Charakter der Erkrankung. Die Bezeichnung rührt ursprünglich daher, dass die Herde eines nicht behandelten Lupus in den Gesichtern der Patienten wie Wolfsbisse aussahen.
Epidemiologie
Die Prävalenz des systemischen Lupus erythematodes ist stark abhängig von der ethnischen Zugehörigkeit. In Europa liegt sie zwischen 25 und 40 Fällen auf 100.000 Einwohner, in Asien beträgt sie rund 50 Fälle pro 100.000 Einwohner. In den USA wurde die Prävalenz mit 178/100.000 Personenjahre und die Inzidenz mit 7,5 pro 100.000 Personenjahre bestimmt.[1] Die größte Prävalenz wird jedoch bei Afroamerikanern und in der Karibik mit etwa 200 Fällen auf 100.000 Einwohner beobachtet. Europaweit liegt das Verhältnis von Frauen zu Männern bei 2,5-3:1. Das Durchschnittsalter für das Auftreten der Erkrankung liegt bei beiden Geschlechtern zwischen 43 und 45 Jahren.
Die chronisch kutane Form des LE macht etwa 2/3 der Fälle aus, die ACLE und SCLE jeweils 1/3 und ICLE 1/10. Durch gleichzeitiges Vorhandensein mehrerer Subtypen ergibt sich ein Wert über 100%.[2] Die 10-Jahres-Überlebensrate liegt bei durchschnittlich 85%.
Einteilung
Der Lupus erythematodes unterteilt sich in verschiedene Unterformen[3]:
- Systemischer Lupus erythematodes (SLE)
- Kutaner Lupus erythematodes (CLE)
- Akut kutaner Lupus erythematodes (ACLE)
- Subakut kutaner Lupus erythematodes (SCLE)
- Chronisch kutaner Lupus erythematodes (CCLE)
- Lupus erythematodes profundus (LEP)
- (Chronischer) Diskoider Lupus erythematodes (DLE bzw. CDLE)
- Chilblain Lupus erythematodes (CHLE)
- Intermittierender kutaner Lupus erythematodes (ICLE)
Eine weitere Form ist der medikamenteninduzierte Lupus erythematodes (DIL), der durch einige Medikamente ausgelöst wird. Das Lupus-ähnliche Syndrom verschwindet für gewöhnlich mit dem Absetzen der Medikamente wieder.
Kinder von Müttern, die an SLE erkrankt sind, können einen Neonatalen Lupus erythematodes (NLE) entwickeln.
Systemischer LE (SLE)
Der SLE betrifft vor allem jüngere Frauen zwischen 20 und 40. Autoantikörper und Immunkomplexe führen zu Gewebeschäden und zum typischen dermatologischen Krankheitsbild. Die SLE ist eine sehr komplexe Erkrankung, die sich sowohl in ihren Symptomen als auch in ihrer Manifestation (bis hin zum Tod) sehr divers zeigt. Als Triggerfaktoren sind eine Reihe von genetischen, hormonellen und Umweltfaktoren bekannt, u.a. UV-Licht, Infektionen, einige Medikamente und eine genetische Prädisposition. Die Erkrankung verläuft über Jahre in Schüben.
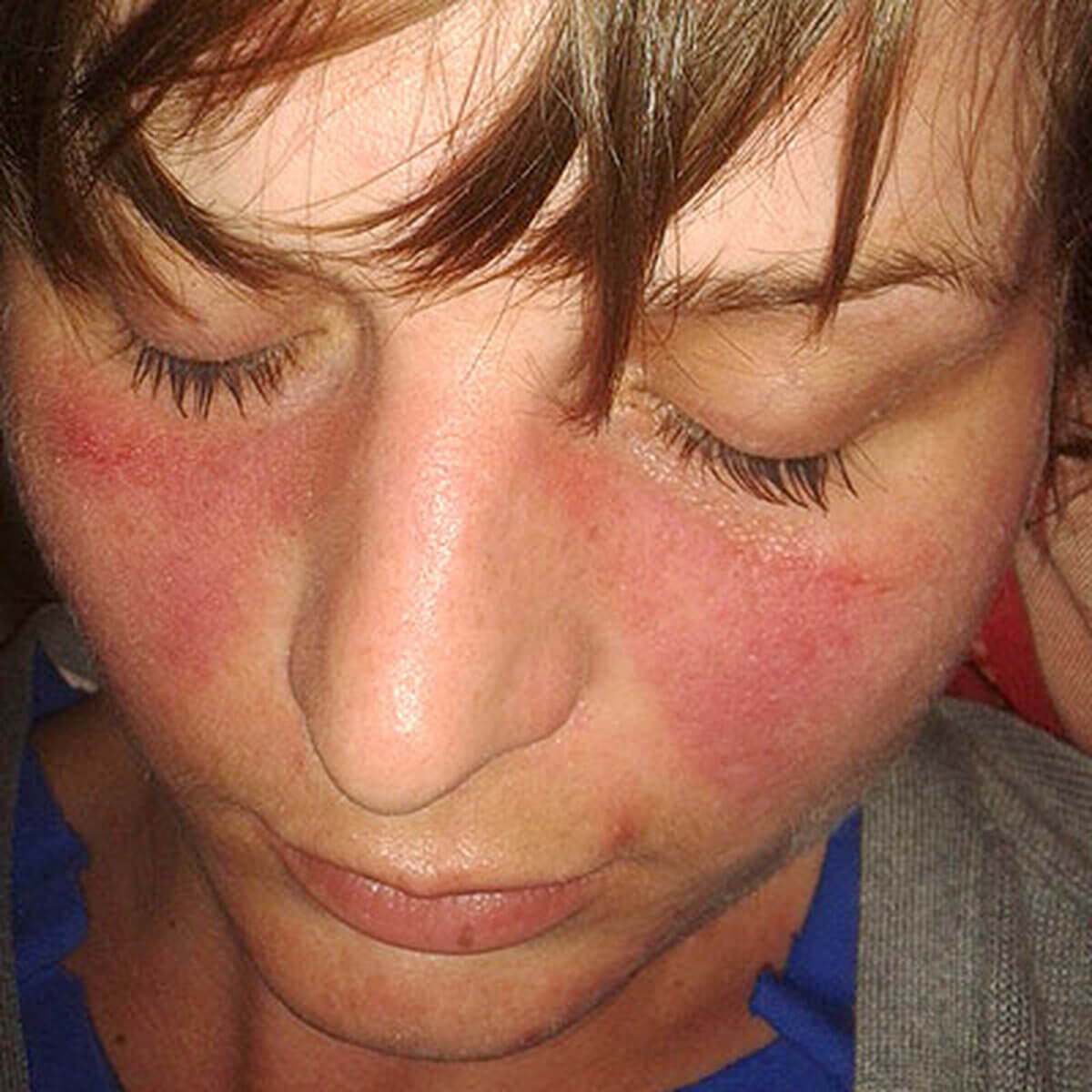
Klinisch findet man das Schmetterlingserythem, makulopapulöse Exantheme an Brust und Rücken, keratotische Plaques an der Dorsalseite der Finger sowie Teleangiektasien und Hämorrhagien am Nagelfalz. Systemisch sind typisch:
- Arthralgien: Es findet sich vor allem eine nicht destruktive Synovialitis der Gelenke mit Auftreten von Fibrin und neutrophilen Granulozyten.
- Pleuritis: Typischerweise zeigt sich eine fibrinöse oder auch mit Erguss einhergehende Entzündung (serofibrinöse Entzündung), die zu Verwachsungen und Fibrosen führen kann.
- Perikarditis
- Endokarditis Libman-Sacks: Atypische verruköse Endokarditis, die grundsätzlich jede Herzklappe betreffen kann. In seltenen Fällen finden sich bei Lupus-Patienten Myokarditiden, die mitunter auch der initial symptomatische Manifestationsort sein können, sodass eine Myokardbiopsie durchgeführt werden sollte.
- Glomerulonephritis
- Pneumonie: Gelegentlich finden sich Lupus-bedingte interstitielle Pneumonien ("Lupus-Pneumonie") oder auch eine Alveolitis. Dabei ist ein Übergang in eine chronische interstitielle Lungenfibrose möglich.
Hinzu tritt oft eine Beteiligung des Nervensystems.
Kutaner LE (CLE)
Bei der kutanen Form des LE ist meistens ausschließlich die Haut betroffen. CLE kann sich aber auch zu einem systemischen LE entwickeln. Die Ätiologie ist nicht vollständig geklärt, neben Rauchen, UV-Licht, Hormonschwankungen und Lebendimpfstoffen, soll auch eine genetische Prädisposition bei der Entstehung eine Rolle spielen.
Die chronisch diskoide Form des LE (DLE/CDLE) verläuft ebenfalls chronisch in Schüben und zeigt sich durch diskoide schuppende rote Plaques mit zentraler Atrophie. Die Disci finden sich hauptsächlich im Gesicht, wo sie als blasse narbige Areale abheilen. Die Berührungsempfindlichkeit ist gesteigert. Eine systemische Beteiligung ist selten. An den Herden sind fest haftende Schuppen zu finden. Entfernt man diese, so bleibt ein zentraler Sporn übrig. Dies bezeichnet man als Tapeziernagelphänomen.


Diagnose
Krankheitskriterien
Das American College of Rheumatology (ACR) hat eine Liste mit elf Kriterien für Lupus definiert. Um die Diagnose sicherzustellen, muss der Patient mindestens vier der folgenden Symptome aufweisen[4]:
- Schmetterlingserythem
- Kutaner Lupus erythematodes: Schuppenartige, verfärbte Hautläsionen
- Photosensitivität: lichtempfindliches Exanthem
- Orale oder nasopharyngeale Schleimhautulzera
- Nephritis mit Proteinurie höher als 0,5 Gramm pro Tag oder Zylindrurie
- Enzephalopathie (Krampfanfälle oder Idiopathische Psychose)
- Nachweis antinukleärer Antikörper (ANA)
- Nachweis von Doppelstrang-DNA-Antikörpern (dsDNA-AK) oder Smith-Antigen-Antikörpern (Sm-AK)
- Pleuritis oder Perikarditis
- Blutbild-Anomalien: hämolytische Anämie, Leukozytopenie, Lymphozytopenie oder Thrombozytopenie
- Nicht-erosive Arthritis
Labordiagnostik
- Immunhistochemie
- Bei Probe-Exzisionen kann mittels Immunfluoreszenz das Lupusband bestimmt werden, das aus IgM, IgG und dem Komplementfaktor C3 besteht.
- Screening
- Erythrozytensedimentationsrate (Blutsenkung, BSG) ↑
- Blutbild mit Differentialblutbild (Autoimmunzytopenie)
- Serumkreatinin
- Urinstatus, Urinsediment
- Antinukleäre Antikörper (ANA), (Fluoreszenzmuster)
- Weitere Untersuchungen bei pos. Screening
- Differenzierung der ANA
- Anti-dsDNA
- Komplementfaktor C3, Komplementfaktor C4
- Antiphospholipid-Antikörper, Lupus-Antikoagulans
- Glomeruläre Filtrationsrate
- Leberenzyme, LDH, bei Muskelbeschwerden auch Creatinkinase
- Weitere Labordiagnostik abhängig von Symptomatik und Komorbidität
Pathohistologie
Der pathohistologische Befund ist abhängig von der Organmanifestation.
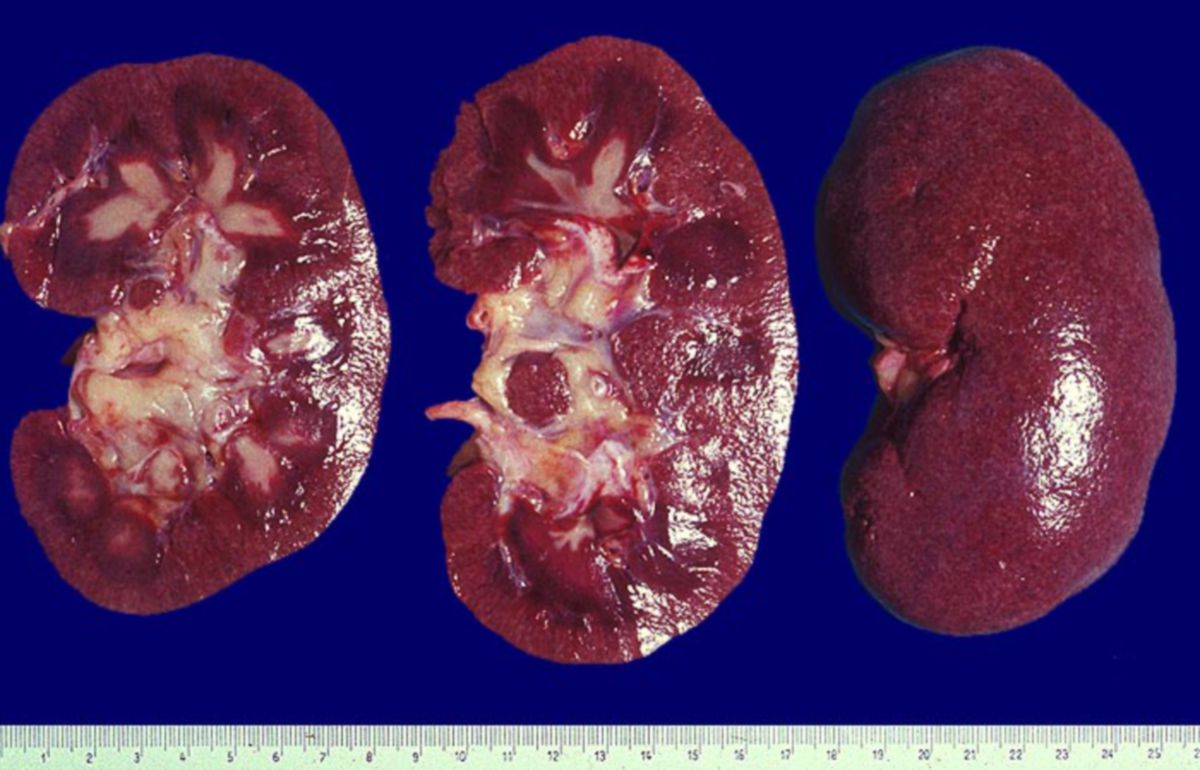
Niere
In etwa 65% der Fälle zeigt sich eine Glomerulonephritis vom Immunkomplextyp im lichtmikroskopischen Präparat. Immunhistochemisch und elektronenmikroskopisch sind in der Regel eine Reihe weiterer morphologischer Veränderungen erkennbar. Funktionell finden sich vermutlich DNA-anti-DNA-Komplexe. Die WHO unterscheidet 5 Reaktionsklassen, welche die Bandbreite an möglichen Veränderungen aufzeigen. Isolierte Präparate ohne Angabe der Symptome können den Pathologen zu einer Fehldiagnose verleiten, weil die Veränderungen auch bei anderen Erkrankungen zu finden sein können.[5]
- Klasse I: normale Niere (sehr selten)
- Klasse II: leichtgradige mesangiale Lupusglomerulonephritis mit mesangialen Ablagerungen von Komplement und Immunglobulin
- Klasse III: fokal-segmentale proliferative Glomerulonephritis, wobei weniger als 50% der Glomeruli betroffen sind
- Klasse IV: diffuse proliferative Glomerulonephritis (der Befund findet sich bei etwa 50% der SLE-Patienten)
- Klasse V: membranöse Glomerulonephritis, zumeist mit schwerer Proteinurie und nephrotischem Syndrom. Eine pathognonomische Morphologie gibt es in dieser Klasse nicht.
Haut
In der Histologie findet sich eine Vaskulitis oder perivaskuläre Infiltrate, zudem Ablagerungen von Immunglobulin und Komplement an der dermoepidermale Grenze. Das in der Immunhistochemie gefundene Lupusband ist bei systemischem Lupus erythematodes üblicherweise überall in der Haut zu finden, bei diskoidem Lupus erythematodes hingegen nur in betroffenen Herden. Der fehlende Nachweis eines Lupusbandes schließt die Erkrankung jedoch nicht aus.
Therapie
Es existiert zur Zeit (2025) keine Heilungsmöglichkeit für Lupus erythematodes. Etwa 25% aller Lupus-Patienten entwickeln nur eine milde Form des SLE mit milder ausgeprägten Hautsymptomen und hämolytischen Symptomen. Diese Patienten werden u.a. behandelt mit:
Schwerwiegendere Fälle erfordern eine intensivere Therapie mit Kortikosteroiden und ggf. Chemotherapie. Immunsuppressiva können während eines Rezidivs oder zur Langzeit-Suppression der Krankheit verabreicht werden.
Die therapeutische Landschaft verändert sich sehr dynamisch. Derzeit (2018) existieren 74 klinische Studien zu Therapien von SLE, die gegen inflammatorische Zytokine, Chemokine und deren Rezeptoren sowie B-Zellen, Plasmazellen, intrazelluläre Signalkaskaden und Interferone gerichtet sind.[6]
Komplikationen
Fortbildung
Literatur
- Kuhn A et al: Diagnostik und Therapie des systemischen Lupus Erythematodes (Übersichtsarbeit), Deutsches Ärzteblatt 2015, frei zugänglich
- Arasteh, Baenkler et al.: Innere Medizin. 2. Auflage, 2009. Thieme Verlag
- Bertsias G, Cervera R, Boumpas DT: Systemic Lupus Erythematosus: Pathogenesis and Clinical Features
- Ziemer M, Milkova L, Kunz M: Lupus erythematosus. Part II: clinical picture, diagnosis and treatment. J Dtsch Dermatol Ges. 2014 Apr;12(4):285-301; quiz 302. doi: 10.1111/ddg.12254.
- Gergianaki I, Bertsias G: Systemic Lupus Erythematosus in Primary Care: An Update and Practical Messages for the General Practitioner. Front Med (Lausanne). 2018 May 29;5:161. doi: 10.3389/fmed.2018.00161.
Quellen
- ↑ Feldman, C.H., Costenbader, K.H.: Epidemiology and classification of systemic lupus erythematosus. In: Rheumatology. Hrsg. Hochberg, M.D., Gravallese et al., 7th. Edition. Elsevier 2019: Printed in China. 1092.
- ↑ Kunz M: Lupus erythematosus. Part I: epidemiology, genetics and immunology. J Dtsch Dermatol Ges. 2013 Aug;11(8):709-19; quiz 720, 709-720; quiz 721. doi: 10.1111/ddg.12165.
- ↑ Leitlinie Kutaner Lupus Erythematodes, 2013
- ↑ How is lupus diagnosed? The American College of Rheumatology
- ↑ Weening JJ et al.: The classification of glomerulonephritis in systemic lupus erythematosus revisited. International Society of Nephrology Working Group on the Classification of Lupus Nephritis; Renal Pathology Society Working Group on the Classification of Lupus Nephritis. Kidney Int. 2004 Feb;65(2):521-30. Review. Erratum in: Kidney Int. 2004 Mar;65(3):1132.
- ↑ Felten R, Dervovic E, Chasset F, Gottenberg JE, Sibilia J, Scher F, Arnaud L.: The 2018 pipeline of targeted therapies under clinical development for Systemic Lupus Erythematosus: a systematic review of trials. Autoimmun Rev. 2018 Jun 6. pii: S1568-9972(18)30133-2. doi: 10.1016/j.autrev.2018.02.011