Komplementsystem










Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenEnglisch: complement system
Definition
Das Komplementsystem ist ein Teil des unspezifischen humoralen Immunsystems, das zur Eliminierung von zellulären Antigenen (z.B. Bakterien) beiträgt.
Übersicht
Das Komplementsystem besteht aus mehr als 40 verschiedenen Proteinen im Blut und auf der Zelloberfläche, die überwiegend in der Leber synthetisiert werden.[1] Diese Proteine werden in Anwesenheit von Pathogenen oder Antikörpern aktiviert.
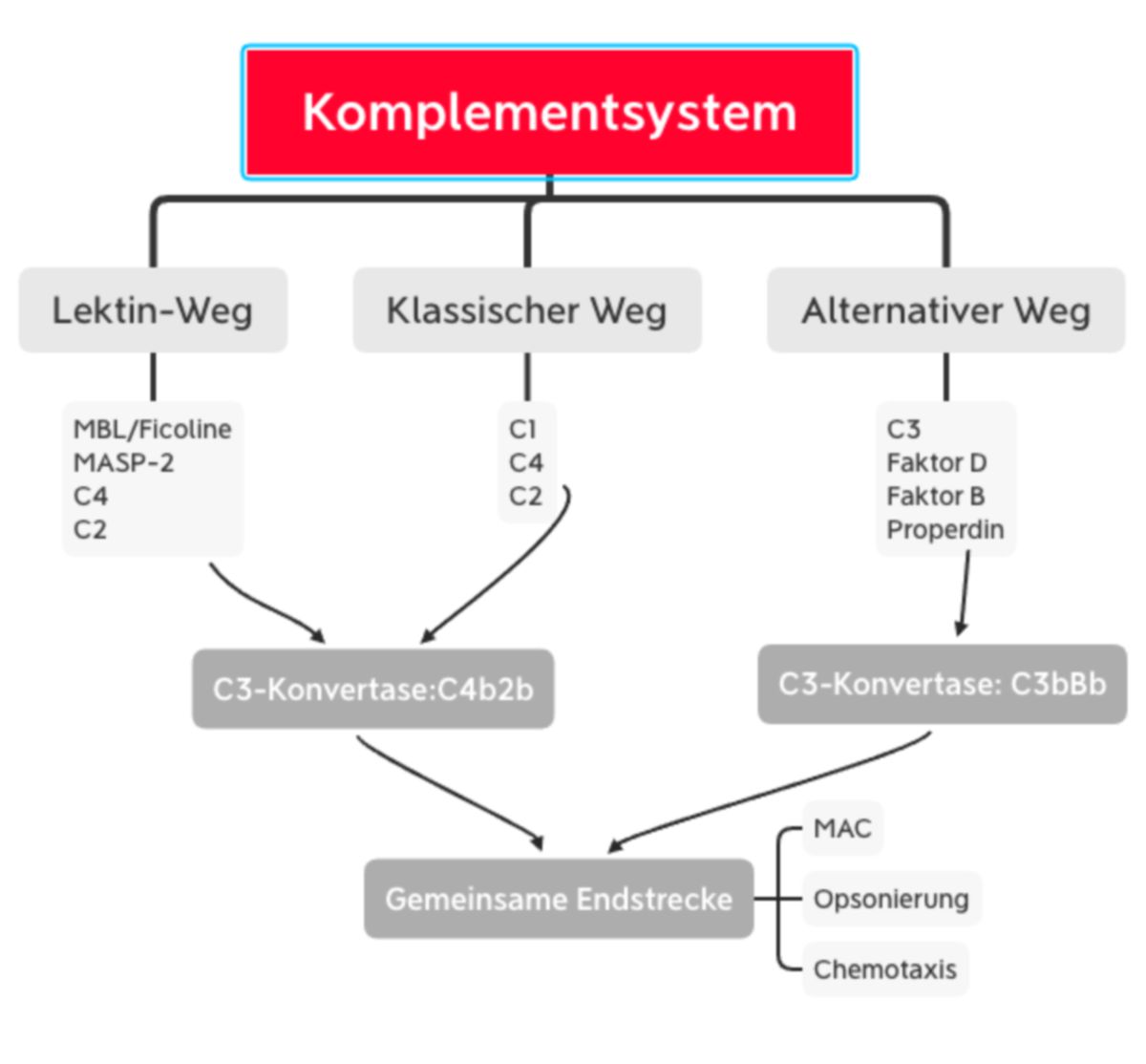
Die meisten der Komplementfaktoren sind Proteasen, die inaktiv als Zymogen synthetisiert werden. Der Aktivierungsmechanismus ist der Blutgerinnung ähnlich: Aktivierte Komplementfaktoren bewirken kaskadenartig eine Proteolyse und damit Aktivierung weiterer Komplementfaktoren. Das Ergebnis der Komplementreaktion ist die Abtötung des Pathogens, entweder direkt durch Bildung eines Membranangriffskomplexes (MAC, lytischer Komplex) oder indirekt durch Stimulation proinflammatorischer Reaktionen (Chemotaxis, Histaminfreisetzung) und Erleichterung der Phagozytose (Opsonierung).
Der MAC ist morphologisch als Pore innerhalb der Zellmembran der zu lysierenden Fremdzelle zu verstehen. Durch Aufhebung der Zellintegrität ermöglicht sie die Destruktion der Zelle.
Neben der Funktion bei der unspezifischen Immunabwehr beeinflusst das Komplementsystem auch das spezifische Immunsystem: Die Opsonierung von Pathogenen durch Komplement erleichtert die Phagozytose durch antigenpräsentierende Zellen, die Komplementrezeptoren (CRs) besitzen. Diese präsentieren anschließend die pathogenen Antigene den T-Zellen. Die B-Zell-Reaktion und das immunologische Gedächtnis werden ebenfalls durch Komplement beeinflusst.[2][3] Außerdem modulieren viele Komplementfragmente die Zytokinproduktion.
Nomenklatur
Die meisten Komplementfaktoren werden mit "C" und einer nachfolgenden Nummer abgekürzt wiedergegeben (z.B. C1). Die Zahl spiegelt nicht die Abfolge der Kaskade wieder, sondern ergibt sich aus der Reihenfolge ihrer Entdeckung:
- Komplementfaktor C1
- Komplementfaktor C2
- Komplementfaktor C3
- Komplementfaktor C4
- Komplementfaktor C5
- Komplementfaktor C6
- Komplementfaktor C7
- Komplementfaktor C8
- Komplementfaktor C9
Spaltprodukte der Komplementfaktoren erhalten einen Buchstaben als Suffix. Dabei erhält das kleinere Fragment den Suffix "a", das größere den Suffix "b". Dabei sind folgende Ausnahmen zu beachten:
- C1: C1q, C1r und C1s sind keine Spaltprodukte, sondern unterschiedliche Proteine, die zusammengefasst als C1 bezeichnet werden.
- Früher wurde bei C2 das größere Fragment als C2a bezeichnet. Inzwischen wird o.g. Konvention auch bei diesem Faktor angewendet.
Weitere Komplementfaktoren, die erst später entdeckt werden, sind z.B. Komplementfaktor B oder Komplementfaktor D. Deren Spaltprodukte erhalten ebenfalls die Sufixe a und b (z.B. Bb).
Bestandteile
| Funktion | Faktor |
|---|---|
| Bindung an Antigen-Antikörper-Komplex und Pathogenoberfläche |
|
| Bindung an Kohlenhydrate auf mikrobieller Oberfläche |
|
| Aktivierung von Enzymen |
|
| Membran-bindende Proteine und Opsonine |
|
| proinflammatorische Mediatoren |
|
| Membranangriffskomplex |
|
| Komplementrezeptoren |
|
| regulatorische Proteine |
|
Komplementkaskade
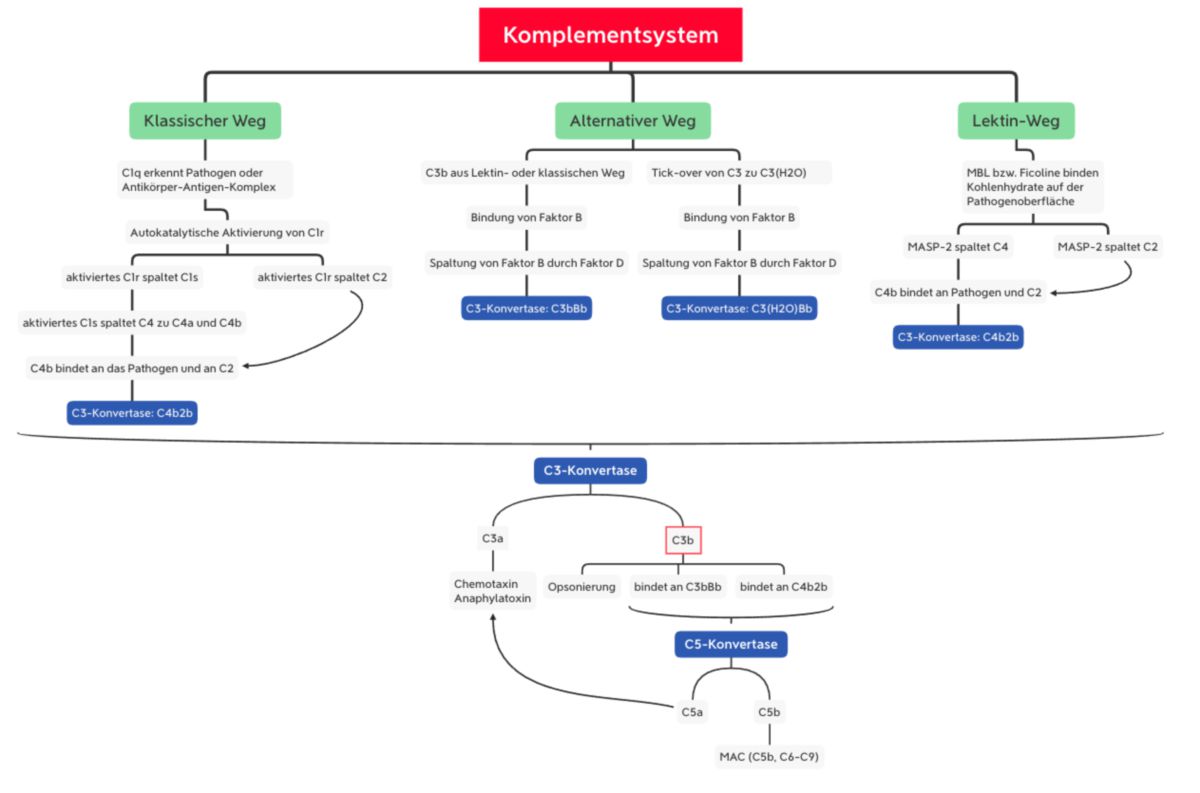
Das Komplementsystem kann auf drei verschiedenen Wegen aktiviert werden:
- Lektinweg: lösliche Kohlenhydrat-bindende Proteine (Mannose-bindendes Lektin und Ficoline) binden an Kohlenhydratstrukturen der mikrobiellen Oberfläche
- klassischer Weg: C1q erkennt eine pathogene Oberfläche oder bindet an Antikörper, die bereits an ein Pathogen gebunden sind. C1q ist mit den Proteasen C1r und C1s assoziiert.
- alternativer Weg: spontane Hydrolyse und Aktivierung von C3, welches an mikrobiellen Oberflächen binden kann.
Alle drei Aktivierungswege enden in der Bildung der C3-Konvertase. Dieser Enzymkomplex kann aus unterschiedlichen Proteinen bestehen, katalysiert jedoch immer den entscheidenden Schritt der Komplementkaskade, die Spaltung von C3 in C3a und C3b.
Während C3a als inflammatorischer Mediator wirkt, unterzieht sich C3b nun einer Konformationsänderung. Dadurch kann die Thioesterbindung von C3b mit Hydroxy- oder Aminogruppen der mikrobiellen Oberfläche reagieren. Die nun entstehende kovalente Bindung an das Pathogen dient der Opsonierung. Außerdem kann C3b an die C3-Konvertase binden und somit die C5-Konvertase bilden. Dieser Enzymkomplex sorgt für die Spaltung von C5:
- C5a wirkt ebenfalls als inflammatorischer Mediator
- C5b bildet mit C6 bis C9 den Membranangriffskomplex
Lektinweg
Viele Mikroorganismen besitzen Kohlenhydratstrukturen, die auf menschlichen Zellen nicht zu finden sind. Diese zu den PAMPs (pathogen-assoziierte molekulare Muster) gehörenden Strukturen werden vom Körper durch folgende, im Blut und extrazellulärer Flüssigkeit zirkulierende Pattern-Recognition-Rezeptoren erkannt:
- Mannose-bindendes Lektin (MBL): oligomeres Akute-Phase-Protein, bindet v.a. Mannose, Fucose und N-Acetylglucosamine (GlcNAc) auf grampositiven und -negativen Bakterien sowie auf Mykobakterien, Hefepilzen und einigen Viruspartikeln und Parasiten.
- Ficoline:
- M-Ficolin (Ficolin-1) wird in der Lunge und von Blutzellen gebildet und sezerniert. Es erkennt acetylierte Zuckerketten wie GlcNaC oder N-Acetylgalactosamine (GalNAc)
- L-Ficolin (Ficolin-2) wird in der Leber gebildet und zirkuliert im Blut. Erkennt acetylierte Zuckerketten (GlcNaC, GalNAc) und Lipoteichonsäure von einigen grampositiven Bakterien.
- H-Ficolin (Ficolin-3) wird in der Leber gebildet und zirkuliert im Blut. Bindet an D-Fucose und Galaktose des grampositiven Bakteriums Aerococcus viridans.
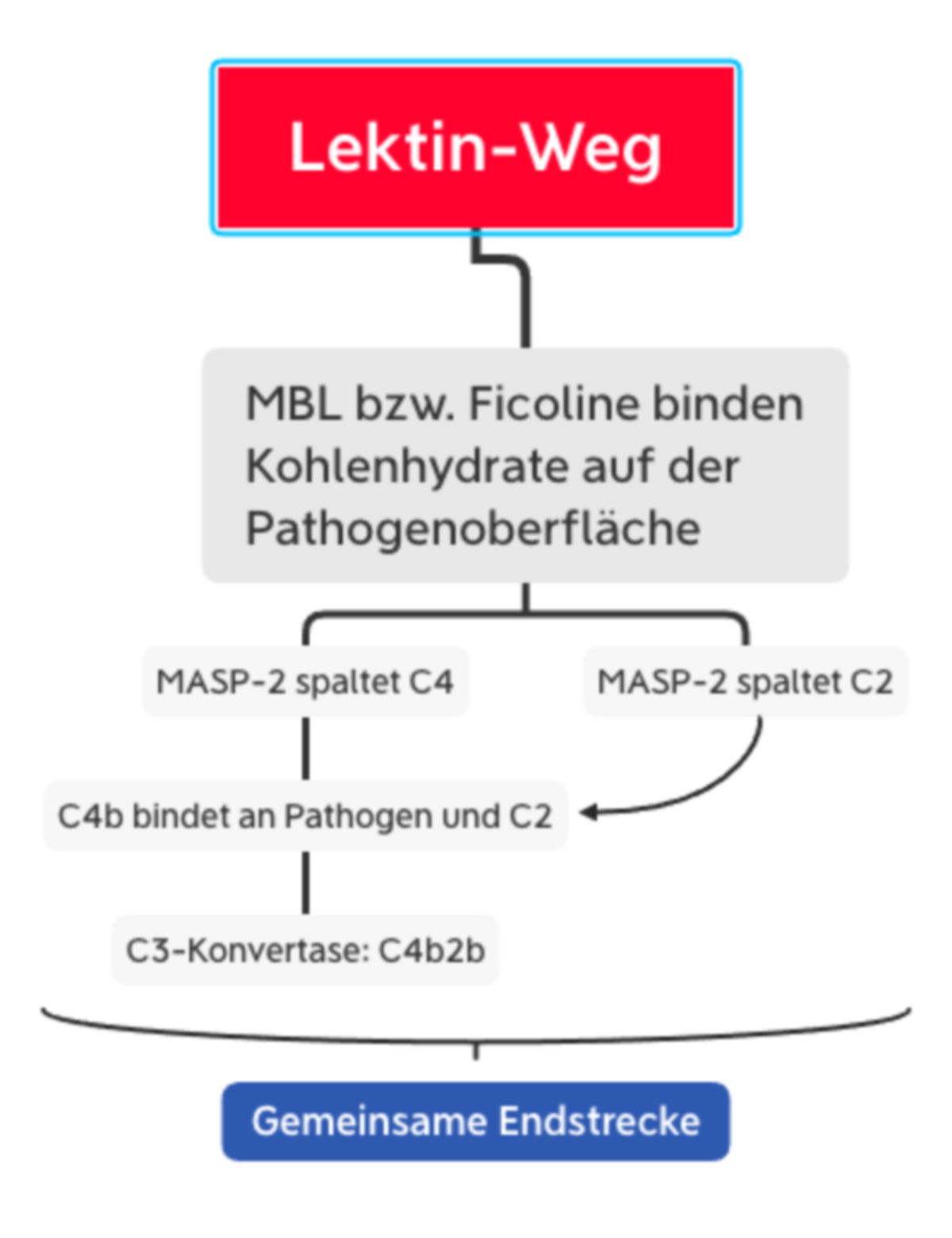
MBL und Ficoline bilden im Plasma einen Komplex mit inaktiven Enzymen, den MBL-assoziierten Serinproteasen MASP1 und MASP2. Wenn MBL bzw. Ficolin an die Pathogenoberfläche bindet, sorgt eine Konformationsänderung in MASP2 für die Aktivierung eines weiteren MASP2-Moleküls. Diese Protease spaltet dann C4 in C4a und C4b. Wie bei C3 besitzt C4 auch eine Thioesterbindung, die nun im C4b-Molekül freiliegt. Das Resultat ist eine kovalente Bindung von C4b an die Pathogenoberfläche, wo es C2 bindet. C2 wird durch die MASP2 gespalten, sodass C2b mit C4b einen Komplex eingehen kann: C4b2b, die C3-Konvertase des Lektinwegs.
Die Funktion von MASP1 ist derzeit (2023) nicht endgültig geklärt.
Klassischer Weg
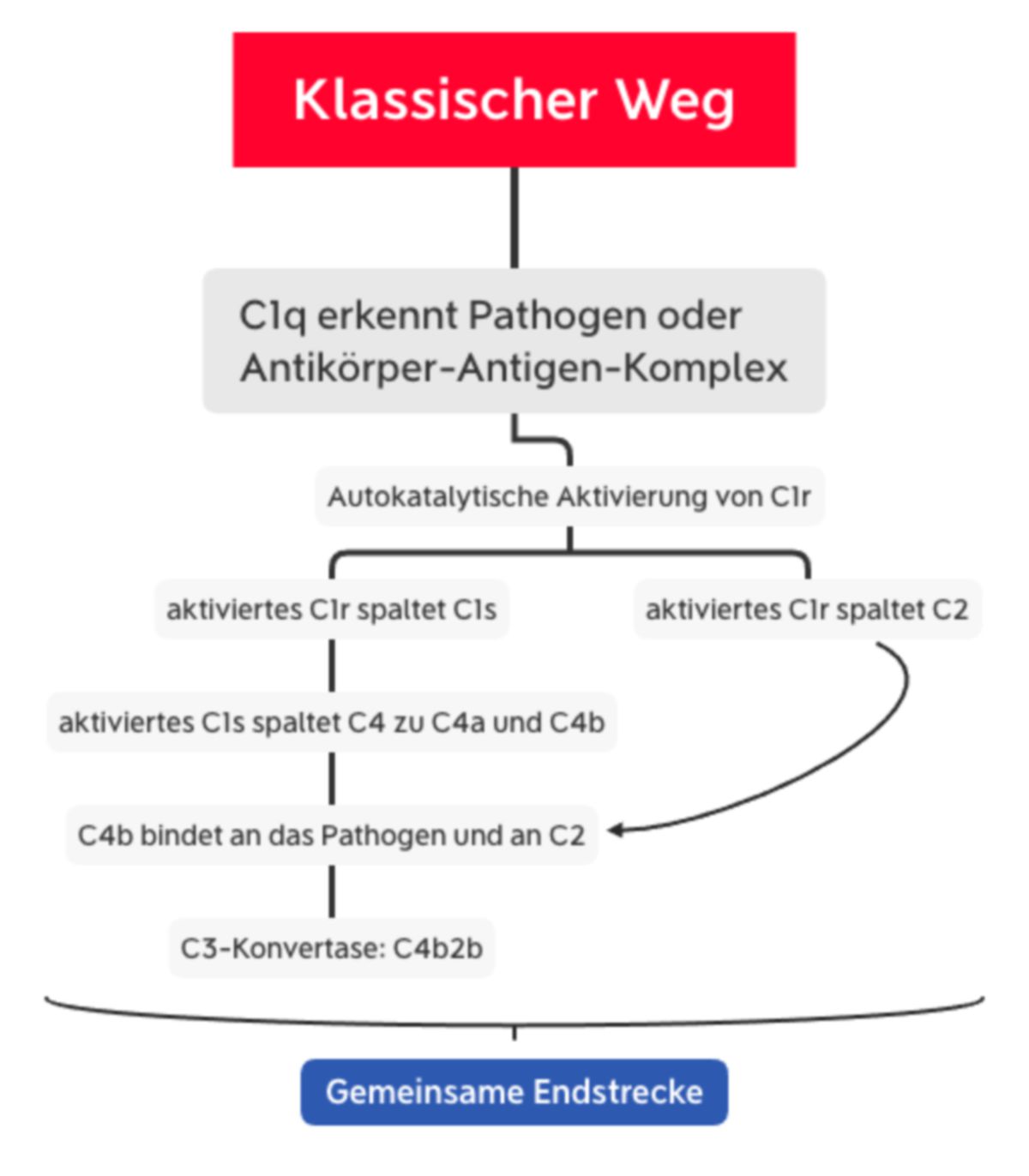
Der klassische Weg ähnelt dem Lektinweg, wobei er statt MBL-MASP-Komplex den C1-Komplex beinhaltet. Dieser besteht aus C1q und den zwei Serinproteasen C1r und C1s.
C1q besteht aus 6 Trimeren, die jeweils eine globuläre Kopfregion besitzen. Diese Region bindet:
- direkt an die Pathogenoberfläche (z.B. an Proteine der Zellwand oder an Lipoteichonsäure),
- an das C-reaktive Protein (CRP), welches an Phosphocholin-haltige Polysaccharide auf der Oberfläche vieler Bakterien bindet,
- oder an die Fc-Region von Antikörpern (v.a. IgM, z.T. IgG), die bereits an ein Antigen gebunden sind.
Wenn mindestens zwei der globulären Köpfe mit dem Liganden interagieren, kommt es zur Konformationsänderung im C1r-C1s-Komplex. Dies führt zur autokatalytischen Aktivierung von C1r, welches dann C1s spaltet. Das nun aktivierte C1s spaltet C4 zu C4a und C4b. C4b bindet C2, das durch C1s in C2a und C2b gespalten wird. Somit entsteht die gleiche C3-Konvertase wie beim Lektinweg (C4b2b).
| Faktor | Funktion |
|---|---|
| C1q | bindet an Pathogenoberfläche oder an Antikörper-Antigen-Komplex |
| C1r | aktiviert C1s |
| C1s | spaltet C4 und C2 |
| C4b | bindet kovalent an das Pathogen (Opsonierung), bindet C2 |
| C4a | schwacher proinflammatorischer Mediator |
| C2a | Präkursor des vasoaktiven C2-Kinin |
| C2b | Bestandteil der C3- und C5-Konvertase zur Spaltung von C3 und C5 |
| C3b | bindet kovalent an das Pathogen (Opsonierung), bindet C5, induziert die Amplifikation auf dem alternativen Weg (s.u.) |
| C3a | proinflammatorischer Mediator |
Alternativer Weg
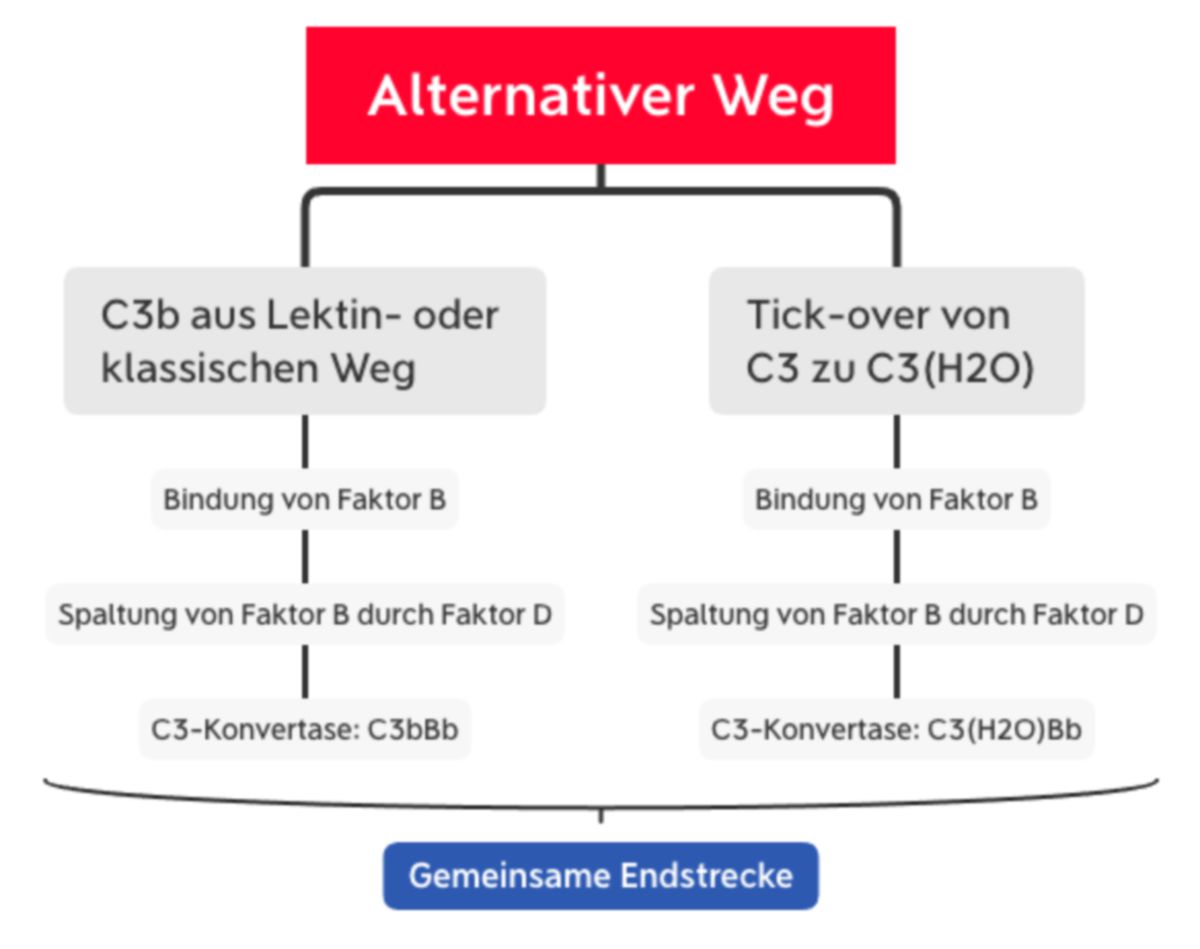
Trotz des Namens ist der alternative Signalweg für ca. 80 % der Komplementaktivierung verantwortlich.[1] Er wird auf zwei Wegen aktiviert:
- durch C3b, das im Lektin- oder im klassischen Weg generiert wird oder
- durch spontane Hydrolyse ("tick-over") der Thioesterbindung von C3. Dadurch entsteht C3(H2O).
C3b bindet den Faktor B und sorgt für seine Konformationsänderung. Dadurch kann Faktor B durch den Faktor D in Ba und Bb gespalten werden. Somit entsteht die C3-Konvertase des alternativen Wegs (C3bBb).
C3(H2O) bindet ebenfalls den Faktor B, der dann durch Faktor D gespalten wird. Dadurch entsteht die sog. "fluid-phase C3-Konvertase" (C3(H2O)Bb). Sie spaltet ebenfalls C3 zu C3a und C3b.[4]
Ingesamt sorgt der alternative Weg also für eine Akkumulation von C3b, das kovalent an das Pathogen bindet und seine Oberfläche bedeckt. Da die C3-Konvertase sehr kurzlebig ist, wird sie durch Properdin (Faktor P) stabilisiert. Properdin wird von neutrophilen Granulozyten produziert und nach Aktivierung freigesetzt.
| Faktor | Funktion |
|---|---|
| C3b | bindet kovalent an das Pathogen (Opsonierung), bindet Faktor B zur Spaltung durch Faktor D, bildet die C3-Konvertase (C3bBb) und C5-Konvertase (C3b2Bb) des alternativen Wegs |
| Ba | unklare Funktion |
| Bb | Bestandteil der C3- und C5-Konvertase |
| D | spaltet Faktor B |
| P | stabilisiert die C3-Konvertase C3bBb |
Endstrecke
Die Bildung der C3-Konvertase (C4b2b im Lektin- und klassischen Weg bzw. C3bBb im alternativen Weg) führt zur Spaltung von C3 in C3b und C3a.
Anschließend kommt es zur Bildung der C5-Konvertase:
- C3b bindet an C4b2b → C4b2b3b
- C3b bindet an C3bBb → C3b2Bb
Die C5-Konvertase bindet C5 und spaltet es in C5b und C5a. Wie bei C3 und C4 beinhaltet C5 eine Thioesterbindung, die nun freigelegt wird. Somit kann C5, wenn auch in geringerem Umfang, an die Pathogenoberfläche binden.
Opsonierung
C3b und in geringerem Ausmaß C4b fungieren als Opsonin, indem sie an Komplementrezeptoren auf Phagozyten binden.
Anaphylatoxine und Chemokine
C4a und C5a binden an spezifische Rezeptoren auf Endothel- und Mastzellen und bewirken eine lokale Entzündungsreaktion. Diese Fragmente werden daher auch Anaphylatoxine bezeichnet. Sie induzieren eine Kontraktion der glatten Muskulatur und eine Erhöhung der Gefäßpermeabilität. Außerdem wird die Bildung von Adhäsionsmolekülen auf Endothelzellen stimuliert und Mastzellen zur Histaminfreisetzung angeregt.
C4a und C5a wirken auch als Chemokine, d.h. sie rekrutieren Phagozyten zum Ort der Infektion. C5a wirkt weiterhin direkt auf Neutrophile und Monozyten und erleichtert die Phagozytose von Pathogenen.
Membranangriffskomplex
Nachdem die C5-Konvertase C5 in C5a und C5b gespalten hat, bindet C5b an C6. Der C5b6-Komplex nimmt anschließend das amphiphile Molekül C7 auf, welches durch eine Konformationsänderung in die Zellmembran eingebaut wird. Der C5b67-Komplex bindet dann C8, das eine Polymerisation von 10 bis 16 C9-Molekülen induziert. Dadurch bildet C9 eine Pore in der Zellmembran mit einer hydrophoben Außenseite und einem hydrophilen Kanal. Der Durchmesser beträgt ca. 10 Nanometer. Das Resultat ist ein Verlust der zellulären Homöostase und die Störung des transmembranären Gradienten. Die Zelle wird somit lysiert.
Regulation
Die dauerhafte Komplementaktivität durch den alternativen Singalweg bietet zwar eine schnelle Immunantwort bei Infektionen, kann aber unreguliert zu Schädigungen des Körpers führen. Daher ist eine Regulation des Komplementsystems zwingend notwendig.
Binden C3b und C4b nicht an die pathogene Oberfläche, wird der Thioester schnell hydrolysiert und damit inaktiviert. Außerdem schützen sich gesunde Zellen vor einer fehlerhaften Aktivität des Komplementsystems durch Oberflächenproteine ("fluid-phase-Regulatoren").[5] Zu den regulatorischen Proteinen gehören u.a.:
- CD55 (decay-accelerating factor, DAF): konkurriert mit Faktor B um die Bindung an C3b an der Zelloberfläche und kann Bb aus der Konvertase entfernen
- Faktor I: spaltet C3b in das inaktive iC3b
- Faktor H: bindet v.a. an C3b, welches fehlerhaft an Wirtszellen gebunden ist; Kofaktor von Faktor I
- CD35 (CR1): hemmt die Bildung der C3-Konvertase, Kofaktor von Faktor I
- C1-Esterase-Inhibitor (C1-INH): entfernt aktiviertes C1r und C1s von C1q, entfernt MASP2 von MBL
- CD59 (Protectin): verhindert die Bildung von Membranangriffskomplexes bei körpereigenen Zellen
Komplementdefekte
Genetische Defekte können Anlass zur Bildung fehlerhafter Komplementfaktoren sein. Komplementdefekte sind für fast jeden Faktor beschrieben. Im Allgemeinen begünstigen sie bakterielle Infektionen, insbesondere mit Kokken. Defizite der terminalen Komplementfaktoren des klassischen Aktivierungsweges (z.B. C9) führen zu einer erhöhten Anfälligkeit für den Genus Neisseria.
Ein spezieller Mangel an C1-Esterase-Inhibitor führt zum hereditären Angioödem.
Im Rahmen eines SLE aktivieren Immunkomplexe das Komplementsystem und haben so einen wesentliche Anteil an der Pathogenese. Auch bei einigen Formen der Glomerulonephritis (z.B. membranoproliferative Glomerulonephritis) tragen fehlgeleitete Aktivierungen des Komplementsystems zum Fortschritt der Erkrankung bei.
Weitere Beispiele für Komplementsystem-vermittelte Erkrankungen sind:
- Paroxysmale nächtliche Hämoglobinurie (PNH)
- Neuromyelitis-optica-Spektrum-Erkrankung (NMOSD)
- generalisierte Myasthenia gravis (gMG)
Diagnostik
Diagnostisch können Komplementdefekte durch den CH50-Test (klassischer Weg) oder den APCH50-Test aufgedeckt werden.
Weblinks
Quellen
- ↑ 1,0 1,1 Merle NS et al. Complement System Part II: Role in Immunity, Front Immunol. 2015;6:257, abgerufen am 14.01.2020
- ↑ Ricklin D et al. Complement: a key system for immune surveillance and homeostasis, Nat Immunol. 2010 Sep;11(9):785-97, abgerufen am 14.01.2020
- ↑ Walport MJ. Complement. First of two parts., N Engl J Med. 2001 Apr 5;344(14):1058-66, abgerufen am 14.01.2020
- ↑ Murphy K. [Janeway's Immunobiology. 8th ed.], New York, NY: Garland Science; 2012:37-73
- ↑ Holmberg MT, et al. Regulation of complement classical pathway by association of C4b-binding protein to the surfaces of SK-OV-3 and Caov-3 ovarian adenocarcinoma cells, J Immunol. 2001 Jul 15;167(2):935-9, abgerufen am 14.01.2020