Myasthenia gravis











Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
Loslegenvon altgriechisch: μῦς ("mŷs") - Muskel, ἀσθένεια ("asthéneia") - Schwäche; von lateinisch: gravis - schwer, ernst
Synonyme: Myasthenia gravis pseudoparalytica, Erb-Goldflam-Krankheit, Erb-Oppenheim-Goldflam-Syndrom
Englisch: myasthenia gravis
Definition
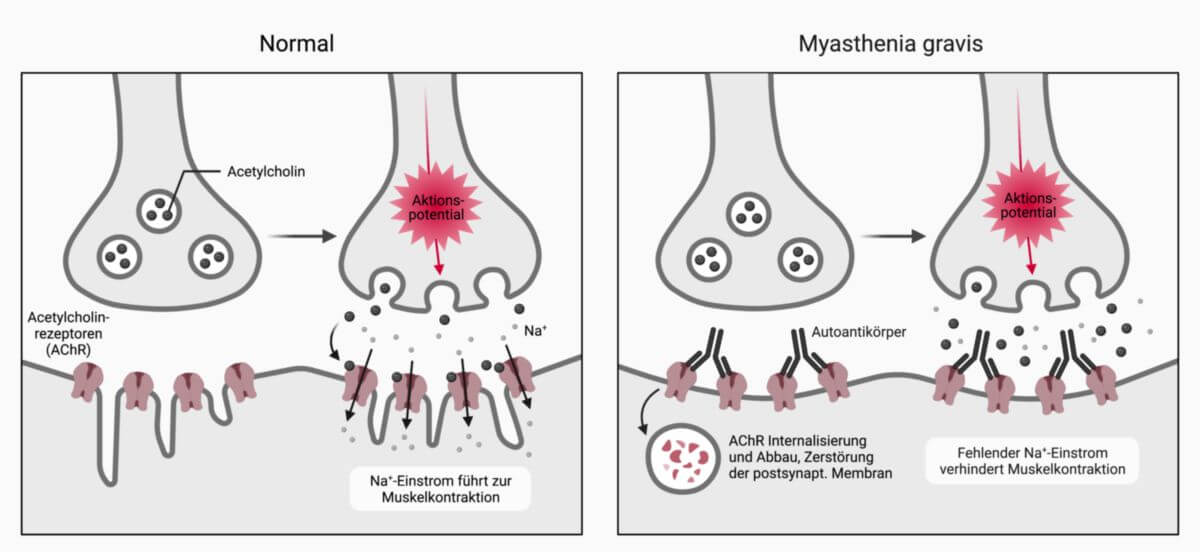
Die Myasthenia gravis ist eine durch Autoantikörper verursachte neuromuskuläre Übertragungsstörung durch Blockierung der Acetylcholinrezeptoren an der motorischen Endplatte (Endplattenerkrankung). Sie führt klinisch zu einer Muskelschwäche, weshalb die Erkrankung von manchen Autoren unter "Muskelerkrankungen" abgehandelt wird.
Pathogenese
Antikörper blockieren die Erregungsübertragung am nikotinergen Acetylcholin-Rezeptor des Skelettmuskels. Die Antikörper lassen sich bei einem Großteil der Patienten mit geeigneten Methoden im Serum nachweisen.
Als Bildungsstätte der Antikörper ist bei den meisten Patienten der Thymus anzusehen. Bis zu 80 % der Patienten weisen eine Thymusveränderung auf: Bei etwa 65 % der Patienten handelt es sich dabei um eine Thymushyperplasie, bei etwa 10 bis 15 % um ein Thymom.
Die zirkulierenden Antikörper binden an den Acetylcholin-Rezeptor und führen durch Aktivierung des Komplementsystems zur Zerstörung der postsynaptischen Membran.
Genetik
Es wurde eine lange Reihe an prädisponierenden HLA-I- und -II-Allelen beschrieben, insbesondere der Allelgruppe HLA-B*08, aber auch HLA-C*07:01, die Allelgruppen DRB1*03, *04, *09, *14, *16, das Allel DRB1*13:02, die Allelgruppe DQB1*02 sowie die Allele DQB1*03:01, *03:02, *03:03, die Allelgruppe HLA-DQB1*05, insbesondere die Einzelallele *05:02 und *05:03, sowie zahlreiche weitere Allele. Einige der Allele sind mit bestimmten Verlaufsformen assoziiert.[1] Andere Allele bzw. Allelgruppen wurden als protektiv beschrieben, z.B. HLA-DRB1*08, DQA1*02, DQB1*06 und DRB1*13.[1]
Epidemiologie
Einteilung
...nach Klinik
Die klinische Klassifikation der aktuellen Leitlinie (Stand 2025) orientiert sich an der Einteilung der MGFA (Myasthenie Gravis Foundation of America), die eine Weiterentwicklung der Ossermann-Klassifikation darstellt.[2]
| MGFA-Klasse | Klinischer Status |
|---|---|
| I | rein okuläre Myasthenie, beschränkt auf äußere Augenmuskeln und Lidschluss |
| II | leichte generalisierte Myasthenie mit Einbeziehung anderer Muskelgruppen, oft einschließlich Augenmuskeln |
| III | mäßiggradige generalisierte Myasthenie, oft einschließlich Augenmuskeln |
| IV | schwere generalisierte Myasthenie |
| V | Intubationsbedürftigkeit mit und ohne Beatmung (myasthene Krise) |
| Die Klassen II bis IV lassen sich in zwei Subgruppen unterteilen: | |
| A | Betonung der Extremitäten und/oder Gliedgürtel, geringe Beteiligung oropharyngealer Muskelgruppen |
| B | besondere Beteiligung oropharyngealer und/oder Atemmuskulatur, geringe oder gleich starke Beteiligung der Extremitäten oder rumpfnaher Muskelgruppen |
...nach Ausbreitung
- Generalisierte Form: Auch wenn die Symptome oftmals im Bereich des Auges beginnen, manifestiert sich das Krankheitsbild hier am gesamten Körper.
- Okuläre Form: Bei dieser Form liegt die Hauptsymptomatik am Auge.
...nach Zeitpunkt des Auftretens
- Juvenile Myasthenia gravis: Hier tritt die Myasthenia gravis bereits im Kindes- bzw. Jugendalter (< 18. Lebensjahr) auf.
- Adulte Myasthenia gravis:
- Myasthenie mit Beginn im frühen Erwachsenenalter (Early-onset Myasthenia gravis, EOMG): Symptombeginn ≤ 50. Lebensjahr, 3 bis 4-mal häufiger bei Frauen
- Altersmyasthenie (Late-onset Myasthenia gravis, LOMG): Symptombeginn > 50. Lebensjahr, häufiger bei Männern
...nach Antikörperstatus[3]
- AChR-Ak-positive MG (ca. 80 %)
- MuSK-Ak-positive MG (ca. 3 %)
- LRP4-Ak-positive MG (ca. 1 %)
- seronegative MG (ca. 15 %)
Symptomatik
Leitsymptom der Myasthenia gravis ist eine belastungsabhängige Muskelschwäche ohne oder mit gering ausgeprägten Muskelschmerzen. Etwa 70 % aller Pat. haben initial eine "okuläre Myasthenie". Diese äußert sich durch Lähmungen der äußeren Augenmuskeln und einer Lidheberschwäche mit Diplopie, asymmetrischer Ptosis und signe de cils („Wimpernzeichen“).[2]
Bei 70–80 % der Patienten generalisiert die Erkrankung.[2] Es kommt zur Schwäche der Gesichtsmuskeln und des Kopfhalteapparates sowie zu Schluck- und Sprechschwierigkeiten. Während die okulären Symptome teilweise zurückgehen, breitet sich die Schwäche auf die proximalen Extremitätenmuskeln aus. Eine Beeinträchtigung der Atemmuskulatur kann zu Belastungs- und Ruhedyspnoe führen.
Bei etwa 10–20 % der Erkrankten kann es zu anhaltenden Spontanremissionen in Form einer erheblichen Besserung der Beschwerden oder einer Komplettremission kommen.
Als Charakteristikum der Myasthenia gravis gilt, dass die Muskelschwäche über den Tag hin zunimmt und sich in Ruhe bessert.
Diagnostik
Klinische Untersuchung
Wichtige Hinweise auf das Vorliegen der Myasthenia gravis gibt die körperliche Untersuchung des Patienten. Betroffene Patienten ermüden bei Durchführung repetitiver Handlungen (z.B. schnelles Öffnen und Schließen der Hand) schnell. Im Simpson-Test zeigt sich bei einem längeren Blick nach oben eine Zunahme der Ptosis.
Im sogenannten Tensilon-Test wird ein kurzwirksamer Acetylcholinesterasehemmer (Edrophoniumchlorid) injiziert. Eine Besserung der Symptomatik ist für die Myasthenia gravis typisch, jedoch nicht spezifisch. Falsch positive und falsch negative Ergebnisse sind nicht selten.
Ebenso ist der Pyridostigmintest möglich, bei dem ebenfalls ein Acetylcholinesterasehemmer verabreicht und beobachtet wird, ob sich dadurch die Symptomatik verbessert.
Elektrophysiologische Untersuchung
In der elektrophysiologischen Diagnostik hat sich die niederfrequente Serienstimulation (2 oder 3 Hz) des motorischen Nervs, der zu einem betroffenen Muskel führt, als geeignet erwiesen. Wenn die 5. Amplitude im Vergleich zur 1. Amplitude mindestens um 10 % kleiner ist, liegt ein für die Myasthenia gravis typisches "Dekrement" vor. Ab der 6. Amplitude folgt ein leichter Wiederanstieg ("U-Shape"), ein geringfügiges "Inkrement". Das pathologische Dekrement ist nicht spezifisch für die Myasthenia gravis.
Labor
Der Antikörpernachweis gegen den Acetylcholinrezeptor im Serum ist aus verschiedenen Gründen nicht immer möglich und ist für die Diagnose einer Myasthenia gravis nicht zwingend erforderlich. Acetylcholinrezeptor-Antikörper (AChR-Ak) finden sich zuweilen auch beim Lambert-Eaton-Syndrom, der amyotrophen Lateralsklerose und bei weiteren Erkrankungen.
Bei einer generalisierten Myasthenie sind bei ca. 80 bis 90 % der Patienten Autoantikörper im Serum nachweisbar. Zum überwiegenden Teil (ca. 90 %) handelt es sich dabei um Acetylcholinrezeptor-Antikörper, die deshalb als erstes gescreent werden. Die polyklonalen AChR-Ak sind gegen unterschiedliche Bereiche des Ionenkanals gerichtet. Unterschieden werden bindende, modifizierende und blockierende AChR-Antikörper. Ein Teil der Antikörper bindet schnell im Endplattenbereich und entgeht der Serumdiagnostik.
Bei seronegativem Laborergebnis (historisch verfestigter Begriff mit der Bedeutung "AChR-Ak-negativ") sollte nach Antikörpern gegen eine skelettmuskelspezifische Rezeptortyrosinkinase (MuSK-Antikörper) gesucht werden. Die MuSK-Antikörper-positive Myasthenie verläuft oft dramatisch mit häufigen myasthenen Krisen.
Bei einem Teil der doppelt seronegativen Myasthenien, d.h. ohne AChR-Antikörper und auch ohne MuSK-Antikörper, wurden in Studien Antikörper gegen das Transmembranprotein Lrp4 gefunden. Das klinische Bild scheint dem der AChR-Antikörper-positiven Myasthenie zu entsprechen.
Mit zunehmendem Alter entwickeln Patienten vermehrt Antikörper gegen Proteine der quergestreiften Muskulatur ("anti-striational antibodies"). Die klinische Bedeutung bedarf noch weiterer Forschung. Einige Antikörper könnten an der Pathogenese der Myasthenia gravis mitwirken.
Titin trägt zur Aufrechterhaltung der Sarkomerstruktur bei. Bei Auftreten von Titin-Antikörpern (besonders gegen das Fragment MGT30) bei Patienten unter 40 Jahren besteht Verdacht auf ein Thymom. Im späteren Alter können Titin-Antikörper auf einen schweren Krankheitsverlauf hinweisen. Anti-Titin-AK können bei Patienten über 60 Jahren allerdings auch ohne Krankheitswert erhöht sein.
Anti-Ryanodin-Rezeptor-Antikörper (RyR-AK) richten sich gegen den Kalziumkanal des Sarkoplasmatischen Retikulums. RyR-Antikörper wurden bei dem Vorliegen von Thymomen gefunden. Die Höhe des Titers entspricht möglicherweise jeweils der Schwere der Erkrankung.
Ein früh einsetzender, krisenhafter Krankheitsverlauf ist auch bei dem Vorhandensein von Antikörpern gegen den spannungsgesteuerten Kalziumkanal Kv1.4 nachgewiesen worden.
Bildgebung
Zur Feststellung von Veränderungen am Thymus ist die Durchführung einer Computertomographie des Thorax sinnvoll, ein vorliegendes Thymom ist aufgrund der möglichen malignen Entartung eine absolute OP-Indikation. Ansonsten ist die Thymektomie bei der okulären Myasthenie umstritten.
Muskelbiopsie
Eine Muskelbiopsie im Endplattenbereich kann im Einzelfall differenzialdiagnostisch angezeigt sein, z.B. zum Ausschluss oder Bestätigung eines kongenitalen Myasthenie-Syndroms.
Differentialdiagnosen
- Myopathien
- Myositiden
- Lambert-Eaton-Myasthenisches-Syndrom
- Mitochondriale Myopathien
- Amyotrophe Lateralsklerose
- Cholinerge Krise
Bei okulärer Myasthenie:
- Erstverdacht zuweilen Multiple Sklerose
- Endokrine Ophthalmopathie
Komplikationen
Bei bis zu 20 % der Erkrankten kommt es zu einer so genannten myasthenen Krise, die durch eine rapide Verschlechterung der Symptomatik gekennzeichnet ist. Sie kann u.a. durch falsch dosierte Arzneistoffe (Betablocker, Acetylcholinesterase-Blocker, Statine[4]), Infektionen, Traumata oder Narkosen sowie ohne erkennbare Ursache ausgelöst werden. Durch Schwächung der Atemmuskulatur kommt es zu einer respiratorischen Insuffizienz mit Aspirationsgefahr. Weitere mögliche Symptome sind:
Das therapeutische Management besteht aus einer intensivmedizinischen, supportiven Therapie und der Suche nach Triggerfaktoren.
Zudem leiden bis zu 80 % der Patienten an einem ausgeprägten, schwer therapierbaren Fatigue-Syndrom.[2]
Verlaufsbeobachtung
Zur Verlaufsbeobachtung der Myasthenia gravis eignen sich verschiedene Selbst- und Fremdbeurteilungsinstrumente. Beispiele sind:
Therapie
Die Therapie der Myasthenia gravis ist multimodal und abhängig von Alter, Thymuspathologie, Antikörperstatus und Krankheitsaktivität. Therapieoptionen sind die symptomatische Therapie, verlaufsmodifizierende Therapien und Interventionstherapien bei Exazerbationen.[2]
Symptomatische Therapie
Rein symptomatische Linderung verschafft die Gabe von Acetylcholinesterasehemmern, wodurch die Acetylcholinwirkung an der postsynaptischen Membran verstärkt werden kann. Eingesetzt wird in der Regel Pyridostigmin, seltener auch Distigmin oder Neostigmin. Dabei ist häufig zu beobachten, dass sich zwar insgesamt die Symptomatik bessert, die typischen klinischen Erscheinungen am Auge jedoch häufig kaum Verbesserung zeigen.
Verlaufsmodifizierende Therapie
Verlaufsmodifizierende Therapieoptionen beinhalten die operative Therapie mittels Thymektomie und die medikamentöse Therapie mit Immunsuppressiva. Gemäß den Leitlinienempfehlungen soll allen Patienten eine verlaufsmodifizierende Therapie angeboten werden.[2]
Thymektomie
Die Thymektomie sollte möglichst als elektive Operation bei klinisch stabilen Patienten durchgeführt werden. Dabei wird ein kausaler Therapieansatz verfolgt – Ziel ist die Elimination der Antikörper-bildenden Strukturen. Die Thymektomie weist Erfolgsraten von etwa 70 % auf, die Besserung der Symptomatik tritt jedoch meistens erst einige Zeit nach der Operation auf.
Indikationen zur Thymektomie sind:[2]
- Nachweis eines Thymoms
- Erwachsene und Kinder ab 13 Jahren mit AChR-Ak-positiver generalisierter Myasthenia gravis (möglichst innerhalb von 2 Jahren bis spätestens 5 Jahren nach Sicherung der Diagnose)
- kann bei seronegativer generalisierter Myasthenia gravis oder LRP4-Ak-positiver generalisierter Myasthenia gravis mit hoher Krankheitsaktivität erwogen werden
- kann bei Kindern und Jugendlichen im Alter von 5 bis 12 Jahren nach Versagen der medikamentösen Therapie erwogen werden
MuSK-Ak-positive Patienten sollen nicht thymektomiert werden.
Verlaufsmodifizierened medikamentöse Stufentherapie
Bei rein okulärer Myasthenia gravis werden Glukokortikoide als als Mittel der ersten Wahl eingesetzt. Alternativ oder in Kombination werden Azathioprin, Mycophenolat-Mofetil, Ciclosporin A oder Methotrexat verwendet (überwiegend Off-Label-Use).
Bei generalisierter Symptomatik empfiehlt die aktuelle Leitlinie folgendes Vorgehen (Stand 2025):[2]
| AChR-Ak positiv | MuSK-Ak positiv | |||
|---|---|---|---|---|
| 1. Wahl | 2. Wahl | 1. Wahl | 2. Wahl | |
| Milder oder moderater Verlauf |
|
|
|
|
| Schwerer Verlauf | Glukokortikoide und/oder ein zusätzliches Präparat aus milder/moderater Verlauf | |||
|
| |||
Die ergänzende Pharmakotherapie besteht hauptsächlich in einer Immunsuppression. Neue Ansätze beinhalten u.a. die Verminderung von IgG durch die Gabe von Efgartigimod alfa für Patienten mit Acetylcholinrezeptor-Antikörpern. Auch andere FcRn-Modulatoren oder Komplementinhibitoren werden (teilweise off-label) eingesetzt. In die Nutzen-Risiko-Abwägung müssen die potenziell schwerwiegenden Nebenwirkungen einbezogen werden.
Interventionstherapien bei Exazerbationen
Im Fall einer drohenden und manifesten myasthenen Krise muss der Patient auf einer Intensivstation behandelt werden. Gegebenenfalls ist eine invasive Beatmung erforderlich. In dieser Situation sind auch intravenöse Immunglobuline oder Plasmapherese bzw. Immunadsorptionsverfahren einzusetzen.[2]
Quiz
Bildquelle
- Bildquelle für Flexikon-Quiz: © Eugene Golovesov / pexels
Quellen
- ↑ 1,0 1,1 Zhong et al., HLA in myasthenia gravis: From superficial correlation to underlying mechanism, Autoimmunity Reviews, 2019.
- ↑ 2,0 2,1 2,2 2,3 2,4 2,5 2,6 2,7 2,8 Wiendl H., Meisel A., Marx A. et al., Diagnostik und Therapie myasthener Syndrome, S2k-Leitlinie, 2024, in: Deutsche Gesellschaft für Neurologie (Hrsg.), Leitlinien für Diagnostik und Therapie in der Neurologie. (abgerufen am 31.07.2025)
- ↑ Berrih-Aknin, S., Frenkian-Cuvelier, M. & Eymard, B. Diagnostic and clinical classification of autoimmune myasthenia gravis. Journal of autoimmunity, 2014
- ↑ Statins: very infrequent reports of myasthenia gravis. MHRA 26.09.2023. Abgerufen am 19.10.2023
Literatur
- Mück A et al. Aktuelle Entwicklungen und neue Therapieoptionen für die Myasthenia gravis. Psychopharmakotherapie 2024
- Leitlinie Diagnostik und Therapie der Myasthenia gravis und des Lambert-Eaton-Syndroms, abgerufen am 03.11.2021