Chronische myeloische Leukämie









Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenSynonyme: chronisch-myeloische Leukämie, CML
Englisch: chronic myeloid leukemia
Definition
Als chronische myeloische Leukämie, kurz CML, wird eine maligne klonale Erkrankung der hämatopoetischen Stammzellen im Knochenmark bezeichnet.
ICD-10-Code
- C92.1 Chronische myeloische Leukämie, BCR/ABL-positiv
- C92.2 Atypische CML, BCR/ABL neg
- C94.8 Blastenkrise bei CML
Einteilung
- Klassische CML: Die klassische CML zählt zu den myeloproliferativen Erkrankungen. Die molekularpathologische Grundlage bildet die BCR-ABL1-Translokation.
- Atypische BCR-ABL1-negative CML: Die atypische CML gehört zu den myelodysplastisch-myeloproliferativen Neoplasien (MDS/MPN). Sie wird inzwischen als MDS/MPN mit Neutrophilie bezeichnet.
siehe Hauptartikel: atypische chronische myeloische Leukämie
Epidemiologie
Die Inzidenz der CML beträgt ungefähr 1,2 - 1,5 pro 100.000 Einwohner und Jahr. In Deutschland erkranken dabei ca. 1.000 - 1.500 Personen pro Jahr. 60% der Patienten sind Männer.
Ein Viertel der Leukämiefälle bei Erwachsenen sind chronisch myeloisch. Die CML kommt zwar in allen Altersgruppen vor, besitzt aber einen Erkrankungsgipfel bei 55 - 65 Jahren. Bei Kindern ist die CML sehr selten. Nur 3 % der CML-Patienten sind unter 20 Jahre alt.
Ätiologie
Die genaue Ursache der chronisch myeloischen Leukämie ist unbekannt. Es liegt eine maligne Entartung der pluripotenten Stammzellen des Knochenmarks vor. Es gibt keine familiäre Häufung und keine eindeutigen Hinweise auf einen Zusammenhang mit einer Exposition gegenüber Toxinen, Insektiziden oder Viren. Die chronische Exposition von Benzol kann jedoch unter bestimmten Voraussetzungen in Deutschland als Berufskrankheit anerkannt werden.
Nur selten tritt die CML sekundär nach Chemo- oder Strahlentherapie auf. Ionisierende Strahlung erhöht dosisabhängig das Risiko mit einem Maximum 5 bis 10 Jahre nach Exposition. Die mediane Zeit bis zum Auftreten einer CML bei Überlebenden der Atombombenabwürfe auf Hiroshima betrug 6,3 Jahre. Nach der Nuklearkatastrophe von Tschernobyl stieg die Inzidenz jedoch nicht an; vermutlich führen nur hohe Dosen zur einer CML.
Pathogenese
Translokation
Bei über 95 % der CML-Patienten liegt eine spezifische Chromosomenaberration vor, die balancierte reziproke Translokation t(9;22)(q34;q11). Sie führt zu einem besonders kurzen Chromosom 22, dem Philadelphia-Chromosom (Ph).
Dabei werden DNA-Sequenzen des Protoonkogens ABL1 (Abelson-Tyrosinkinase) von Chromosom 9 in die Region des Breakpoint Cluster Region-Gens (BCR) auf Chromosom 22 eingefügt. Das BCR-ABL-Fusionsgen kodiert nun für das onkogene Fusionsprotein BCR-ABL1, eine konstitutiv aktive Tyrosinkinase. Diese bewirkt eine exzessive Proliferation und eine reduzierte Apoptose der CML-Zellen. Im Verlauf kommt es zur Suppression der physiologischen Hämatopoese. Normale Stammzellen können aber persistieren und nach effektiver Therapie wieder aktiv werden.
Die Translokation kann in myeloischen Zellen (incl. erythrozytären Zellen, Megakaryozyten und Monozyten), seltener in reifen B- und T-Zellen, jedoch nicht in Stromazellen des Knochenmarks oder in anderen Körperzellen nachgewiesen werden.
Zwar ist ein kausaler Zusammenhang von BCR-ABL1-Translokation und Entstehung von CML in experimentellen Modellen nachgewiesen. Jedoch können BCR-ABL1-Transkripte auch bei gesunden Personen gefunden werden. Daraus lässt sich schließen, dass BCR-ABL alleine keine CML auslöst. Vermutlich sind weitere molekulare Ereignisse oder eine fehlerhafte Immunerkennung von Zellen mit BCR-ABL notwendig.
Varianten
Es existieren verschiedene Mutationen des Fusionsgens, die auch eine therapeutische Relevanz besitzen. Je nach Molekulargewicht unterscheidet man drei Typen des Fusionsproteins. P210 ist die häufigste Form und wird von den Transkripten e13a2 und e14a2 kodiert.
Wenn eine kürzere BCR-Sequenz mit ABL1 fusioniert, entsteht das kleinere BCR-ABL1-Onkoprotein p190 (Transkript e1a2). Es findet sich nur selten bei einer CML und ist dann mit einer schlechteren Prognose verbunden. P190 kommt aber bei zwei Drittel der Patienten mit Ph-positiven akuter lymphatischer Leukämie (ALL) vor.
Die dritte Form p230 wird durch das Transkript e19a2 kodiert und ist mit einem indolenten Verlauf assoziiert.
Pathophysiologie
Die konstitutive Aktivierung von BCR-ABL1 führt zur Autophosphorylierung und Aktivierung zahlreicher Downstream-Signalwege, die unter anderem die Transkription und die Apoptose beeinflussen. Dabei bestehen zahlreiche Interaktionen z.B. mit Ras/Raf/MAPK-, JAK-STAT-, PI3K/Akt-Signalweg oder MYC. Das Verständnis dieser Signaltransduktionswege ist entscheidend für die Entwicklung neuer therapeutischer Ansätze.
Die Abhängigkeit der Tumorzellen von der BCR-ABL1-Aktivierung wird auch als "Oncogene Addiction" bezeichnet.
Langfristig führt die BCR-ABL-Tyrosinkinase zu einer genomischen Instabilität der leukämischen Stammzelle. Folglich entstehen weitere Chromosomenaberrationen und Mutationen, z.B. Ph1-Duplikation, Isochromosom 17, Trisomie 8, Trisomie 19 oder Mutation in p53. Nach einigen Jahre entwickelt sich eine Dominanz des Zellklons, welcher das Philadelphia-Chromosom aufweist. Die normale Hämatopoese wird dadurch sukzessive unterdrückt.
Verlauf
Vor Einsatz spezifischer Therapieformen zeigte sich bei den meisten Patienten typischerweise ein dreistufiger Verlauf. Die vorherrschenden Symptome hängen insbesondere von der Verfügbarkeit und dem Zugang zur Gesundheitsversorgung ab.
In Deutschland wird die CML bei 50 – 60 % der Patienten zufällig im Rahmen routinemäßiger Blutuntersuchung gestellt. In Ländern mit schlechter Gesundheitsversorgung wird die CML erst spät bei Vorliegen von Symptomen bzw. Komplikationen diagnostiziert.
Die einzelnen Stadien können nach WHO-Definition 2016 oder anhand der Kriterien des European Leukemia Net (ELN) definiert werden.[1][2]
Chronische stabile Phase
Die CML beginnt mit der chronischen stabilen Phase (CP). Diese dauert 6 Monate bis 20 Jahre. Mehr als 97% der erwachsenen Patienten befinden sich bei der Erstdiagnose in der CP. Bei Kindern ist dieser Anteil mit 92 bis 95% geringer, hier befinden sich 5 bis 8% der Patienten bei der Erstmanifestation bereits in einer fortgeschrittenen Krankheitsphase.
Per definitionem finden sich im peripheren Blut ≤ 2 % und im Knochenmark ≤ 5 % Blasten.
Akzelerationsphase
Die Akzelerationsphase (AP) bezeichnet den Übergang zwischen der chronischen Phase und dem Blastenschub. Ursächlich sind vermutlich weitere chromosomale Veränderungen. Laut Definition der ELN liegt eine Akzelerationsphase bei Vorliegen von mindestens einem der folgenden Kriterien vor:
- 15 - 29 % Blasten in Blut oder Knochenmark
- Blasten und Promyelozyten im Blut oder Knochenmark insgesamt über 30 % (mit < 30 % Blasten)
- mindestens 20 % Basophile in Blut oder Knochenmark
- therapieunabhängige Thrombopenie < 100.000/µl
- Thrombozytose > 1.000.000/µl
- neu entstandene klonale Evolution
- progrediente Knochenmarkfibrose
- therapierefraktäre progrediente Splenomegalie und ansteigende Leukozytenzahl
Die WHO setzt die Akzelerationsphase bei Vorliegen von mindestens einem der folgenden Kriterien an:
- persistierende oder ansteigende Leukozytose > 10.000/µl trotz Therapie
- persistierende oder ansteigende Splenomegalie trotz Therapie
- persistierende Thrombozytose > 1.000.000/µl trotz Therapie
- persistierende Thrombopenie < 100.000/µl, therapieunabhängig
- > 20 % Basophile im peripheren Blut
- 10 - 19 % Blasten im peripheren Blut und/oder Knochenmark
- zytogenetische Zusatzaberrationen in Philadelphia-Chromosom-positiven Zellen zur (z.B. Isochromosom 17)
- jegliche neue klonale chromosomale Aberration einer Philadelphia-Chromosom-positiven Zelle unter Therapie
- hämatologische Resistenz gegenüber dem ersten Tyrosinkinaseinhibitor (TKI) oder kein komplettes hämatologisches Ansprechen auf den ersten TKI
- jegliche hämatologische, zytogenetische oder molekulare Hinweise auf eine Resistenz gegenüber zwei sequenziell verabreichten TKIs
- Auftreten von zwei oder mehr BCR-ABL1-Mutationen unter Therapie
Blastenkrise
Ohne Therapie kommt es typischerweise 3 bis 5 Jahre nach Erstdiagnose zur Blastenkrise. Dabei liegen nach WHO-Definition über 20 % Blasten, nach ELN-Einteilung über 30 % Blasten im Blut oder Knochenmark vor.
Ein weiteres diagnostisches Kriterium sowohl nach WHO als auch nach ELN ist der Nachweis einer Proliferation extramedullärer Blasten (z.B. in der Haut, im Weichteilgewebe oder in Form einer Osteolyse).
Der Verlauf der Blastenkrise ähnelt einer akuten Leukämie und endet unbehandelt rasch letal.
Dabei kommt es in zwei Drittel der Fälle zu einer myeloischen (selten erythrozytär, promyeloisch, monozytär, megakaryozytär), in einem Drittel zu einer lymphatischen Blastenkrise. Die Unterscheidung hat eine therapeutische Konsequenz, da die lymphathische Blastenkrise auf eine Chemotherapie nach ALL-Protokoll (Cyclophosphamid, Vincristin, Doxorubicin, Dexamethason) in Kombination mit einem TKI ansprechen kann.
Klinisches Bild
Initial sind 50 % der Patienten in der chronisch stabilen Phase asymptomatisch. Je weiter die Erkrankung voranschreitet, desto eher treten die Symptome auf. Leitsymptom der CML ist eine Leukozytose mit kontinuierlicher Linksverschiebung. Weitere Symptome bzw. Befunde sind unter anderem:
- Unspezifische Allgemeinsymptome: Allgemeines Krankheitsgefühl, B-Symptomatik (Gewichtsverlust, Fieber, Nachtschweiß), Müdigkeit, Leistungsminderung
- Splenomegalie: Entsteht durch Infiltration der roten Milzpulpa durch reife und unreife Granulozyten; kann zu Druckgefühl im linken Oberbauch und frühem Sättigungsgefühl führen
- Anämie
- Thrombozytose oder seltener Thrombopenie
- Basophilie: zeigt vor allem den Übergang in die Akzelerationsphase an
- Klopf- oder Druckschmerz im Bereich des Sternums
Da die exzessiv produzierten Granulozyten funktionstüchtig sind, kommt es im Gegensatz zur akuten Leukämie initial nicht zu rezidivierenden Infekten. In der Akzelerationsphase finden sich aufgrund der Zunahme unreifer BCR-ABL-positiver Blasten mit geringerem Differenzierungsvermögen immer weniger funktionsfähige reife Granulozyten. Dadurch steigt das Infektrisiko.
Komplikationen
Komplikationen entstehen durch die veränderte Zahl oder Funktion der peripheren Blutzellen. Eine Thrombozytose führt zu einem erhöhten Risiko für Thrombosen, eine Thrombopenie zu einer erhöhten Blutungsneigung. Außerdem kann durch Bindung des von-Willebrand-Faktors an Thrombozyten ein erworbenes von-Willebrand-Syndrom entstehen.
Da die Granulozyten und ihre Vorstufen gut verformbar sind, kommt es nur selten zu rheologischen Problemen. Bei extremer Leukozytose sind leukämische Thromben jedoch möglich. Entsprechende Manifestationen sind:
Diagnostik
Anamnese
Anamnestisch sollte nach oben genannten Symptomen gefragt werden, z.B. Abgeschlagenheit, Schwäche, Appetitlosigkeit, Gewichtsverlust, Knochenschmerzen, Oberbauchschmerzen.
Körperliche Untersuchung
Der häufigste körperliche Befund bei einer CML ist die Splenomegalie, die bei 20 - 70 % der Patienten vorzufinden ist. Eine Hepatomegalie findet sich in 10 bis 20 % der Fälle. Selten entstehen als Folge der extramedullären Blutbildung kutane bzw. subkutane Läsionen, die Blasten enthalten. Letzteres ist ein Kriterium, welches eine Blastenkrise definiert. Eher untypisch ist eine Lymphadenopathie, die nur in ca. 5 - 10 % der Fälle vorkommt.
Weitere körperlichen Befunde entstehen durch erwähnte Komplikationen, z.B. Sehstörungen bei Zentralvenenthrombose. Eine hohe Basophilie kann zur Histaminüberproduktion mit Pruritus, Diarrhoe und Hitzewallungen führen.
Blutuntersuchung
Bei Verdacht auf eine CML sollte ein Differentialblutbild angefordert werden. Dabei fallen folgenden Befunde auf:
- Anämie: Bei etwa 33 - 80 % der Patienten tritt eine meist milde Anämie auf.
- Leukozytose insbesondere durch Vermehrung neutrophiler Granulozyten: Im Gegensatz zu anderen Formen der Leukämie tritt bei der CML immer eine erhebliche Erhöhung der Leukozyten auf. Die CML weist sogar die höchsten Leukozytenzahlen von allen Leukämien auf, zum Teil mit Werten über 500.000/µl. Hohe Leukozytenzahlen sind unter Umständen schon in Form einer breiten Leukozytenmanschette bei der Blutsenkung erkennbar.
- Linksverschiebung: Es finden sich Leukozyten mit Dominanz von Neutrophilen und Vorhandensein aller Reifestufungen bis zum Myeloblasten (im Gegensatz zur leukämoiden Reaktion oder akuten Leukämie)
- Basophilie, Eosinophilie: Häufig ist ein vermehrtes Auftreten von basophilen und eosinophilen Granulozyten zu beobachten.
- Insbesondere intial zeigt sich in 50 % der Fälle eine erhöhte Thrombozytenzahl (Thrombozytose), in 10% der Fälle eine verminderte Thrombozytenzahl (Thrombozytopenie), meist liegt auch eine Funktionsstörung der Plättchen (Thrombozytopathie) vor.
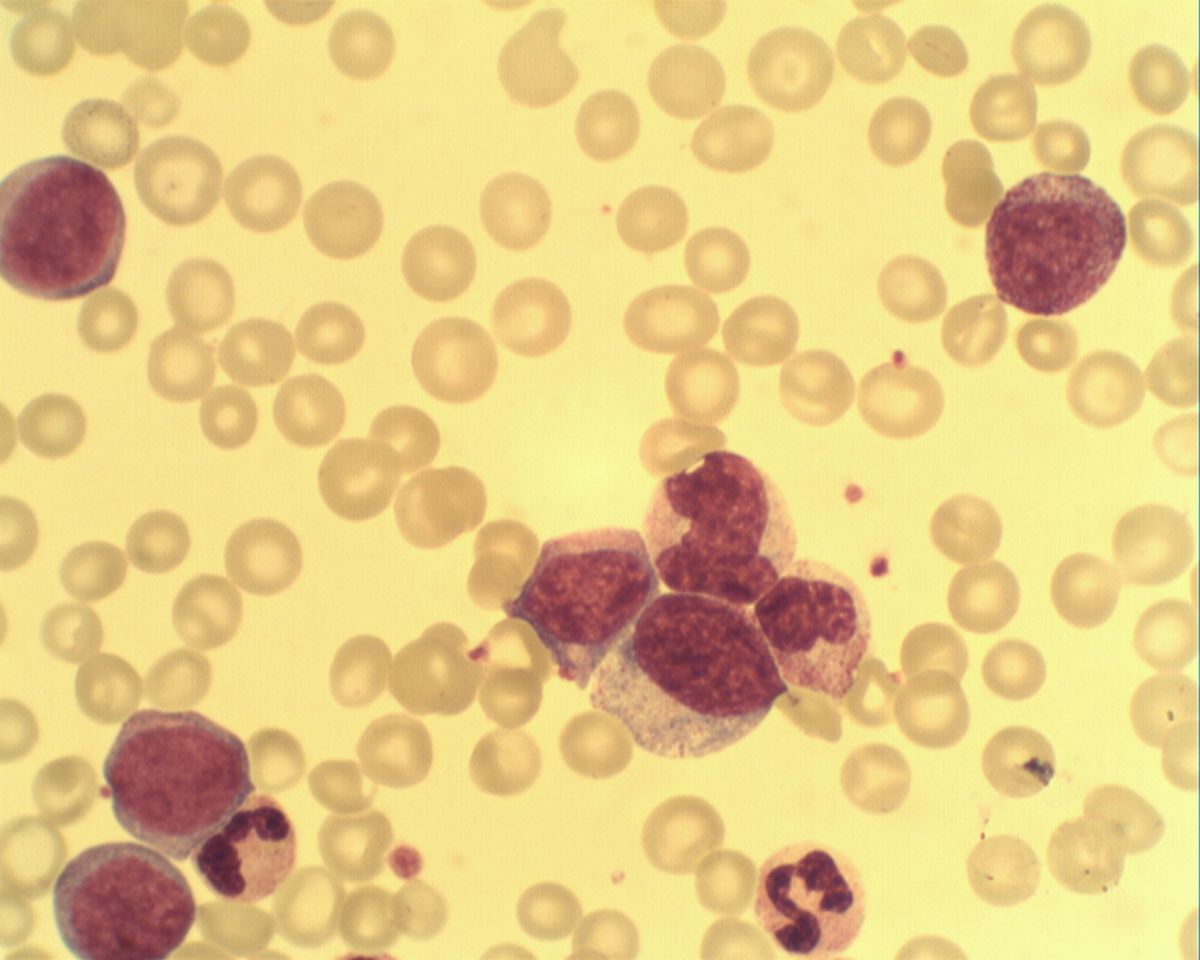
Im Verlauf entsteht eine Myelofibrose mit Verdrängung der normalen Hämatopoese. Folglich kommt es zur extramedullären Blutbildung z.B. in der Milz. Dabei können kernhaltige rote Vorstufen im peripheren Blut auftreten.
Weiterhin findet man erhöhte Plasmakonzentration von Vitamin B12, Harnsäure, LDH und Lysozym. Die BSG ist typischerweise erhöht.
Ein zytogenetischer Nachweis von BCR-ABL-Transkripten erfolgt per PCR.
Knochenmarkuntersuchung
Im Knochenmarkaspirat können zytologische, zytogenetische und zytochemische Untersuchungen durchgeführt werden:
- Zytologie: Hyperzellularität, dominierende Granulopoese (G-E-Index erhöht), kleine Megakaryozyten, Pseudo-Gaucher-Zellen oder meerblaue Histiozyten, Basophilie, Eosinophilie, meist unter 5 % Blasten (chronische Phase)
- Zytogenetik: Metaphasen-Analyse mit Nachweis des BCR-ABL-Fusionsgens bzw. Philadelphia-Chromosoms
- Zytochemie: Die Aktivität der alkalischen Leukozytenphosphatase (ALP) ist stark vermindert. Dies unterscheidet die CML von allen anderen myeloproliferativen Erkrankungen.
Das Knochenmarkbiopsat wird histologisch untersucht. Neben einer Fibrose zeigt sich eine Vermehrung der Promyelozyten und Myelozyten.
Zytogenetische-molekularbiologische Untersuchung
Bei der klassischen CML kann das Philadelphia-Chromosom und das BCR-ABL-Fusionsgen mittels FISH oder RT-PCR nachgewiesen werden. Der Nachweis des Philadelphia-Chromosoms bzw. von BCR-ABL-Transkripten sichert dabei Diagnose. Die Quantifizierung von BCR-ABL in Blut und Knochenmark dient auch dem Monitoring der Therapie.
Einige Patienten haben komplexe Translokationen (variantes Ph-Chromosom), bei denen weitere Chromosomen transloziert sind. Andere Patienten haben ein maskiertes Ph-Chromosom mit Translokation zwischen Chromosom 9 und einem anderen Chromosom als Chromosom 22. In diesen Fällen ist die Prognose und Therapie vergleichbar mit der klassischen CML.
Außerdem besitzen 5 bis 10 % der Ph-positiven Zellen weitere Aberrationen (z.B. Trisomie 8, Isochromosom 18, 17p-Deletion).
Immunphänotypisierung
Die Immunphänotypisierung dient der Charakterisierung der Blasten, insbesondere während der Blastenkrise. Dies hat eine Relevanz für das Ansprechen auf die Therapie und für die Prognose.
Radiologie
In der Bildgebung fällt bei den meisten Patienten eine Splenomegalie auf. Weiterhin zeigt sich eine diffuse Knochenmarkinfiltration – beispielsweise durch ein abnormal erniedrigtes T1w-Signal in der MRT.
Differenzialdiagnosen
- Myeloische leukämoide Reaktion: Unter anderem bei chronischen eitrigen Infektionen, Sepsis oder Gabe von G-CSF kann es zur Leukozytose kommen. Dabei bleibt die Leukozytenzahl meist unter 100.000/µl. Im peripheren Blut treten Granulozyten mit sogenannter toxischer Granulation auf, jedoch keine Basophilie und nur sehr selten Myeloblasten
- Lymphatische leukämoide Reaktion: hohe Lymphozytenzahlen kommen bei einigen viralen Infekten oder Pertussis vor
- Chronische myelomonozytäre Leukämie: Ph-negativ, ALP bei Typ I und II erhöht
- andere myeloproliferative Erkrankungen, z.B. chronische Neutrophilenleukämie (CSF3R-Mutation)
- Primäre Myelofibrose: Splenomegalie, Leukozytose mit Linksverschiebung, Thrombozytose, ALP erhöht
- Atypische CML: CSF3R- oder SETBP1-Mutationen, dysplastische Granulozytopoese (hyposegmentierte Pseudo-Pelger-Zellen)
- Myelodysplastisches Syndrom (MDS): Im Gegensatz zum MDS zeigen die peripheren Blutzellen bei CML nur wenige Dysplasiezeichen
- ALL: 20 % der erwachsenen und 5 % der pädiatrischen Patienten besitzen ein Philadelphia-Chromosom
- Akute myeloische Leukämie (AML): Unter 2 % aller Patienten besitzen ein Philadelphia-Chromosom. Bei der AML liegt aufgrund eines Mangels an Zwischenstufen im peripheren Blut der sogenannte Hiatus leucaemicus vor
Therapie
siehe Hauptartikel: Therapie der chronischen myeloischen Leukämie
Chronische Phase
Nach Sicherung der Diagnose sollte die Therapie in hämatologischen Zentren unter Teilnahme an kontrollierten klinischen Studien stattfinden. Das Ziel ist der Therapie ist eine weitgehende Normalisierung des Blutbildes im Sinne einer Remission. Dabei werden drei verschiedene Formen der Remission unterschieden:
- hämatologische Remission (HR)
- Vollständige hämatologische Remission (CHR, complete hematologic remission)
- Gute hämatologische Remission (MHR, major hematologic remission)
- zytogenetische Remission (CyR)
- Vollständige zytogenetische Remission (CCyR, complete cytogenetic remission)
- Gute zytogenetische Remission (MCyR, major cytogenetic remission)
- molekulare Remission (MR)
- Vollständige molekulare Remission (CMR, complete molecular remission)
- Gute molekulare Remission (MMR, major molecular remission)
Zur Erstlinientherapie sind Tyrosinkinaseinhibitoren empfohlen. Dabei werden abhängig von Alter, Geschlecht, Komorbiditäten und möglichen Nebenwirkungen Imatinib, Nilotinib, Dasatinib oder Bosutinib verwendet. Die TKIs führen in ungefähr 60 bis 80 % zu einer molekularen Remission. Die 10-Jahres-Überlebensrate unter Therapie beträgt 80 bis 90 %.
Es existieren definierte Kriterien für ein Therapieansprechen: Im optimalen Fall ist die Lebenserwartung fast normal, bei Versagen des Therapieansprechens sollte rasch eine Mutationsanalyse und eine Zweitlinientherapie erfolgen. Dabei kommen ein bisher nicht verwendeter TKI oder Ponatinib in Frage.
Grundsätzlich muss in allen Stadien die allogene Stammzelltransplantation (SZT) als potentiell kurative Behandlung erwogen werden. Die 10-Jahres-Überlebensrate nach allogener SZT beträgt ungefähr 55 %.
Akzelerationsphase
Bei Patienten, die sich in der Akzelerationsphase befinden und vorher bereits mit einem TKI vorbehandelt wurden, wird je nach Mutationsstatus ein alternativer TKI verwendet.
Bei Patienten ohne Vorbehandlung kommen ebenfalls Imatinib, Bosutinib, Dasatinib und Nilotinib sowie bei geeigneten Patienten und Spenderverfügbarkeit eine allogene Stammzelltransplantation in Frage.
Blastenkrise
Bei einer myeloischen Blastenkrise helfen Hydroxyurea, Cytarabin und ggf. Anthrazykline, während bei der lymphatischen Form unter anderem Vincristin und Dexamethason eingesetzt werden.
Weiterhin können wie bei der Akzelerationsphase alle TKIs (außer Nilotinib) sowie eine allogene SZT erwogen werden.
Prognose
Vor Einführung der TKIs lag die mediane Überlebenszeit unter Behandlung mit Hydroxyurea, Busulfan und Interferon-alpha bei 3 bis 7 Jahren, die 10-Jahres-Überlebensrate bei 30 %. Die jährliche Mortalität in ersten beiden Jahren lag bei 10 %, anschließend bei 15 bis 20 %.
Inzwischen (2019) kann eine allogene SZT zur Heilung führen, während TKIs selbst bei Vorliegen einer CMR vermutlich nicht kurativ sind, da ganz frühe leukämische Stammzellen nicht ausgelöscht werden. Jedoch führen TKIs meist zu einer fast normalen Lebenserwartung. In diesem Sinne kann die CML als indolente Erkrankung angesehen werden.
Für die klinische Einschätzung der Prognose existieren verschiedene Scores:[3]
- Sokal-Score : basiert auf klinischen und labormedizinischen Daten
- EURO-Score nach Sokal und Hasford: basiert auf Daten vor Einführung der TKI-Therapie
- EUTOS-Score: nutzt den Anteil der Basophilen im peripheren Blut und die Milzgröße zum Diagnosezeitpunkt für die Vorsage des Erreichens einer CCyR
- ELTS-Score: aktuell (2019) empfohlener Prognosescore
Literatur
- Harrisons Innere Medizin Suttorp N, Möckel M, Siegmund B et al.; 19. Auflage. 2016; ABW Wissenschaftsverlag
- Herold, G.: Innere Medizin 2019. Köln: Gerd Herold, 2018
- Onkopedia Leitlinie, Stand Juni 2018, abgerufen am 16.09.2019
Quellen
- ↑ Arber DA et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia, Blood 2016 127:2391-2405, abgerufen am 16.09.2019
- ↑ ELN, abgerufen am 16.09.2019
- ↑ CML-Scores, abgerufen am 16.09.2019