Pankreaskarzinom











Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenSynonym: Bauchspeicheldrüsenkrebs
Englisch: pancreatic carcinoma
Definition
Das Pankreaskarzinom ist ein aus dem exokrinen Anteil des Pankreas entstehendes Karzinom. Es ist abzugrenzen von selteneren Neoplasien des endokrinen Pankreas wie beispielsweise dem Insulinom.
Epidemiologie
In Deutschland erkranken 10 von 100.000 Einwohnern im Jahr neu (Inzidenz) an einem Pankreaskarzinom. In den USA ist das Pankreaskarzinom die fünfthäufigste krebsbedingte Todesursache. Der Altersgipfel für das Auftreten des Pankreaskarzinoms liegt zwischen dem 6. und 8. Lebensjahrzehnt.
Ätiologie
Die Ätiologie des Pankreaskarzinoms ist weitgehend unbekannt. Bei einem kleinen Teil der Patienten findet sich ein familiäres Pankreaskarzinom. Bisher bekannte Risikofaktoren für die Entstehung eines Pankreaskarzinoms sind:
Demographische Risikofaktoren
- Alter (zuverlässigster und wichtigster Risikofaktor)
- Geschlecht (Männer häufiger betroffen als Frauen)
- ethnische Herkunft (höchste Mortalität in der schwarzen Bevölkerung)
Genetische Risikofaktoren und Grunderkrankungen
- familiäres Auftreten von Pankreaskarzinomen
- hereditäre Pankreatitis
- hereditäres non-polypöses kolorektales Karzinom
- Ataxia-Teleangiektasie-Syndrom
- familiäres Mammakarzinom
- familiäres atypisches Nävi-Syndrom
- chronische Pankreatitis
- Diabetes mellitus
- Zustand nach Gastrektomie
Umwelt- und Lifestyle-Risikofaktoren
- Alkoholabusus[1]
- Nikotinabusus
- Exposition gegenüber aromatischen Aminen (Nitrosamine)
- berufliche Exposition (nur geringe Evidenz vorhanden)
Spezifische Diätempfehlungen zur Risikoreduktion gibt es auch nach der aktuellen Leitlinie nicht (Stand 2024). Auf übermäßigen Alkoholkonsum und jeglichen Tabakkonsum sollte jedoch verzichtet werden. Regelmäßige körperliche Aktivität wird empfohlen.
Die Pathogenese ist derzeit (Stand 2024) nicht vollständig verstanden. Die Erkenntnisse deuten auf eine klassische Karzinogenese hin, bei der durch Mutationen in krebsassoziierten Genen schrittweise die Karzinomentstehung begünstigt wird. Beispielsweise sind in über 90 % der Fälle Mutationen im KRAS-Gen und CDKN2A nachzuweisen. Weiterhin ist in über der Hälfte der Patienten das ERBB2-Protoonkogen amplifiziert (wie beim Mammakarzinom).
Pathologie
Pankreaskarzinome betreffen in 60 % der Fälle den Pankreaskopf, in 10 % der Fälle den Corpusbereich, in weiteren 10 % der Fälle den Pankreasschwanz. Bei 20 % ist das gesamte Organ diffus betroffen.
Fast alle Karzinome des Pankreas sind Adenokarzinome, die aus den Epithelien des Pankreasganges hervorgehen, sogenannte duktale Adenokarzinome des Pankreas (PDAC). Sie rufen in der Regel eine desmoplastische Reaktion hervor. Dadurch erscheint der Tumor makroskopisch grau und hart. Bereits in frühen Stadien infiltriert das Pankreaskarzinom das umliegende Gewebe und unter Umständen bereits anliegende Strukturen.
Pankreastumoren im Kopfbereich komprimieren die Ampulla hepatopancreatica, den gemeinsamen Ausführungstrakt für Galle und das exokrine Sekret des Pankreas. Dadurch kommt es zur Cholestase mit Erweiterung der Gallengänge. Eine weitere oft beobachtete Eigenschaft von Pankreaskarzinomen im Kopfbereich ist die Invasion in das umgebende Duodenum.
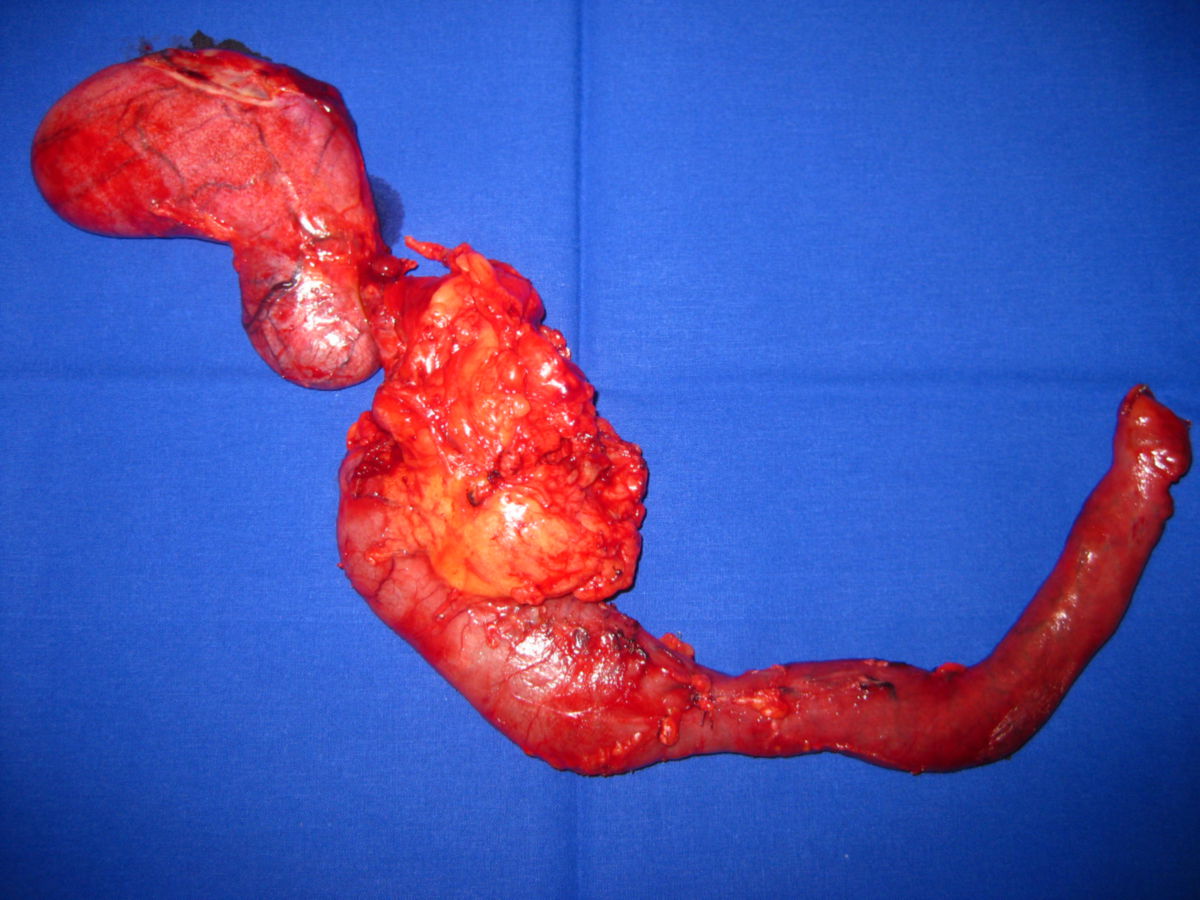
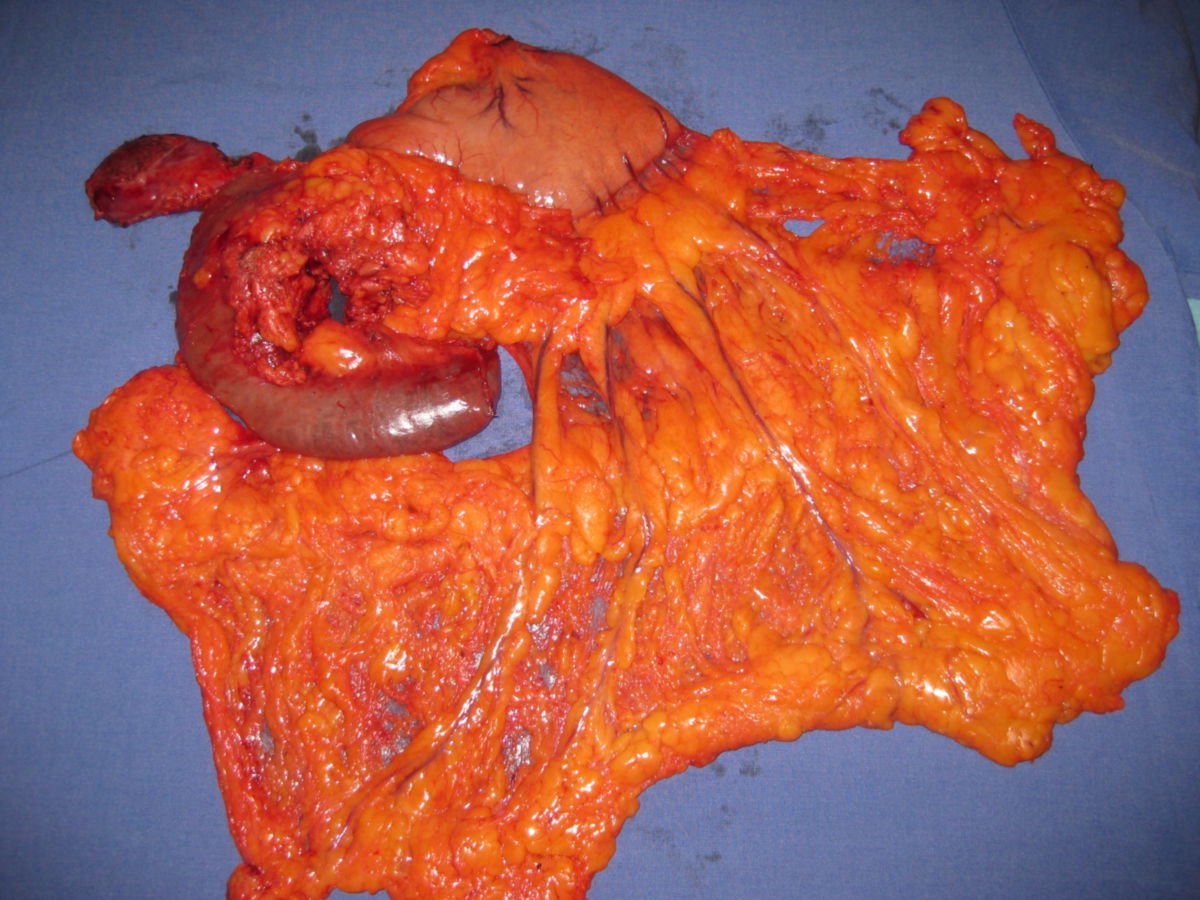
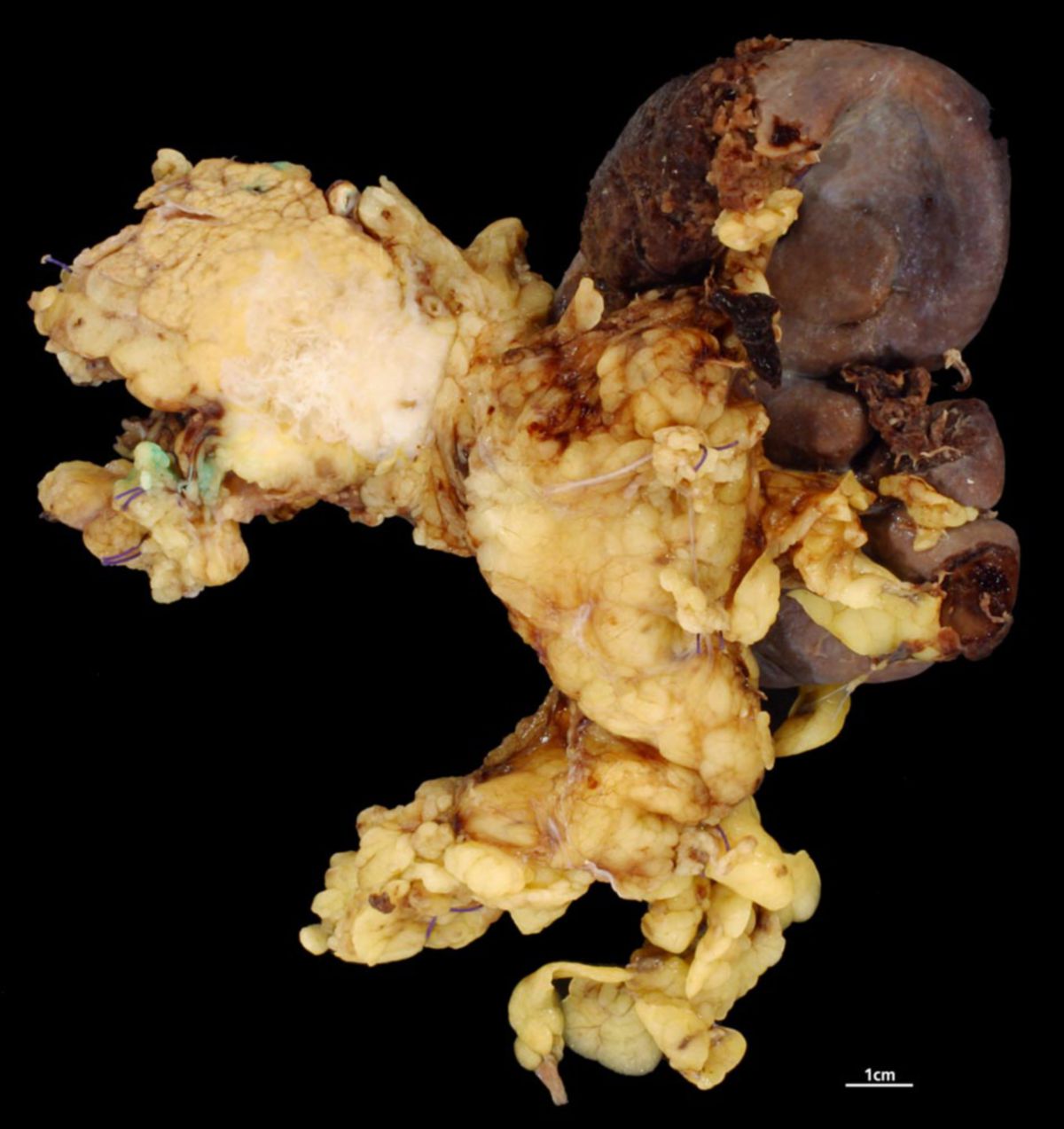
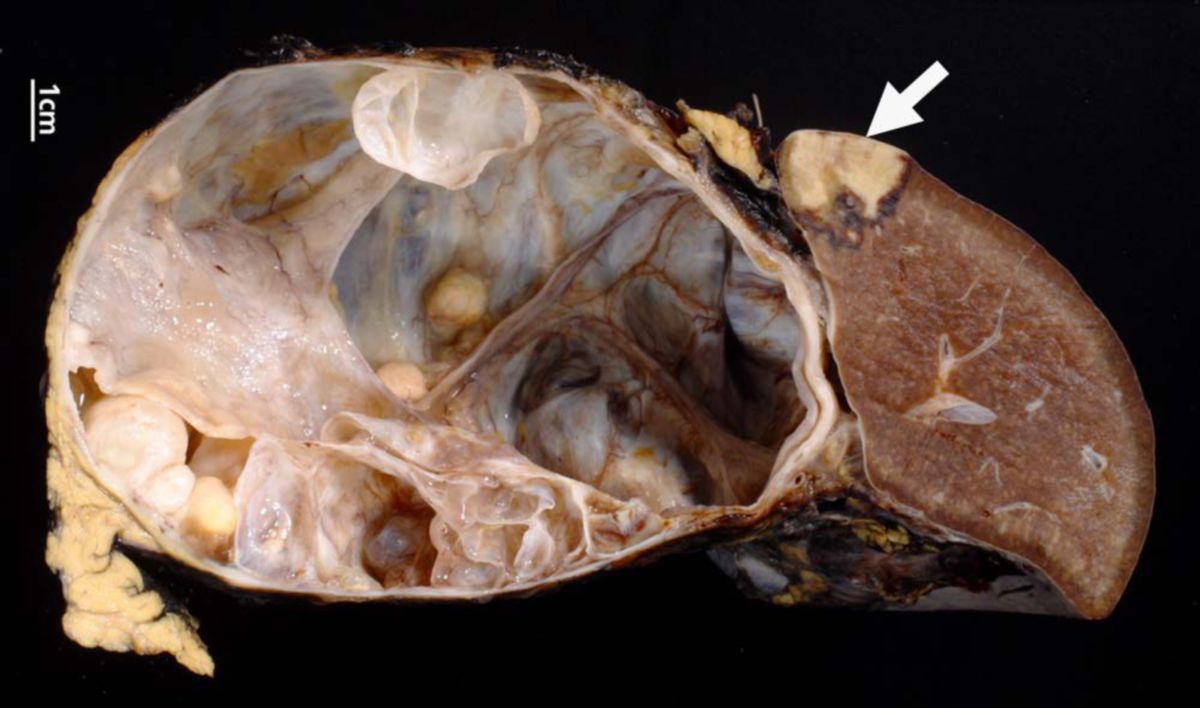
Im Gegensatz hierzu verhalten sich Pankreaskarzinome des Corpus und des Schwanzbereichs für längere Zeit still. Eine Cholestase bleibt aus. Stattdessen infiltriert das Pankreaskarzinom das Retroperitoneum, befällt anliegende Organe (Nerven, Milz, Nebenniere, Wirbelsäule, Colon, Magen, Leber und die in diesen Bereichen befindlichen Lymphknoten).
Fernmetastasen treten bei beiden beschriebenen Arten des Pankreaskarzinoms relativ frühzeitig auf. Betroffen sind hiervon hauptsächlich Lungen und Knochen.
Pathohistologie
Das Pankreaskarzinom wächst meistens in glandulärer Form. Bei Muzinsekretion spricht man von muzinösen Pankreaskarzinomen. Die Drüsen des Karzinoms sind unregelmäßig und atypisch. Das sie überziehende Epithel ist aus anaplastischen Zellen zusammengesetzt. Seltener wächst das Pankreaskarzinom völlig undifferenziert.
Adenokarzinom des Pankreas (duktal). HE-Färbung. Histologiepräparat freundlicherweise zur Verfügung gestellt durch die Pathologie der Uniklinik Köln
Molekularpathologie
Mutierte Onkogene, die beim Pankreaskarzinom eine Rolle spielen:
Symptome
Das Pankreaskarzinom ist oft lange Zeit asymptomatisch. Symptome ergeben sich erst dann, wenn benachbarte Strukturen infiltriert und verdrängt werden. Aufgrund der späten Symptomatik bestehen in ca. 80 % der Fälle bei Diagnosestellung bereits Metastasen. Daher ist bei verdächtiger Symptomatik eine schnelle Abklärung angezeigt.
Durch die retroperitoneale Lage des Pankreas, frontal der Wirbelsäule, macht sich ein Pankreaskarzinom zu Beginn der Erkrankung häufig durch Rückenschmerzen bemerkbar. Weitere häufige Symptome eines Pankreaskarzinoms sind:
- Oberbauchschmerzen
- Störungen der Verdauung
- Übelkeit
- Appetit- und Gewichtsverlust
- Ikterus
- positives Courvoisier-Zeichen
In wechselndem Ausmaß kann es zu einer Steatorrhö (bei Cholestase) und in Folge der Zerstörung von Inselzellen zu einem Diabetes mellitus kommen. Durch den Druck auf die umgebenden Gefäße wird in einem Teil der Fälle eine Milzvenenthrombose ausgelöst. Die systemische Wirkung freigesetzter Pankreasenzyme auf die Blutgerinnung kann auch Thrombosen in entfernt gelegenen Venen, z.B. in den Beinvenen auslösen (Trousseau-Syndrom).
Diagnostik
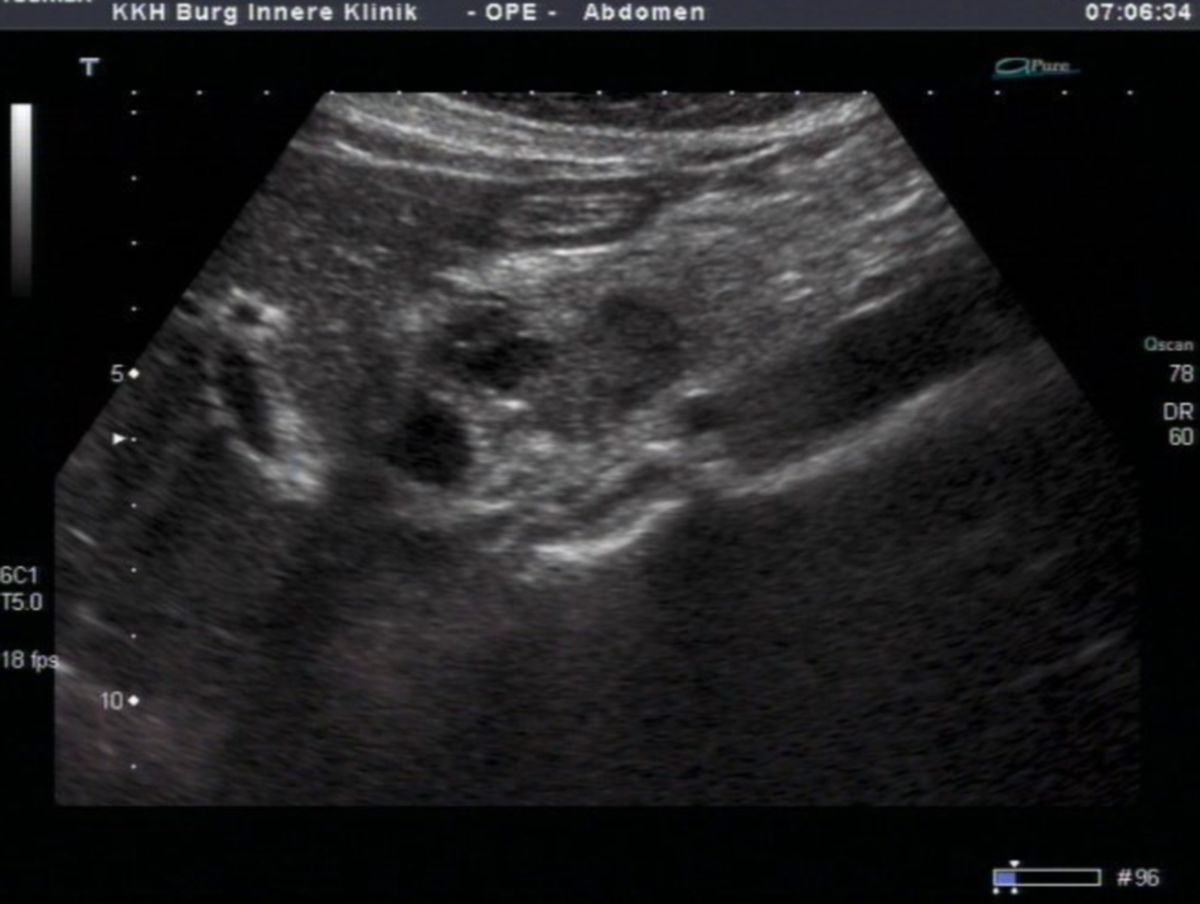
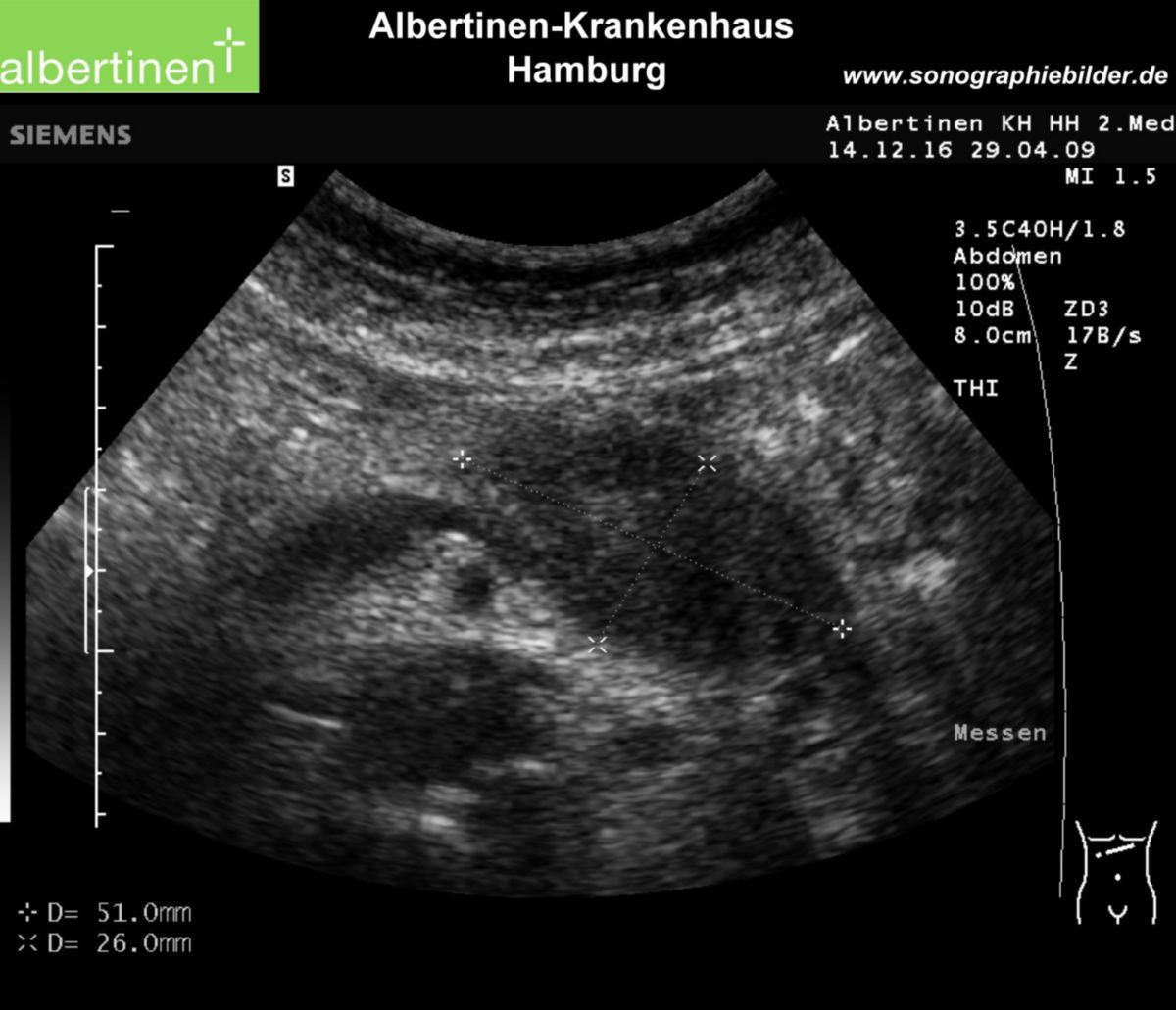
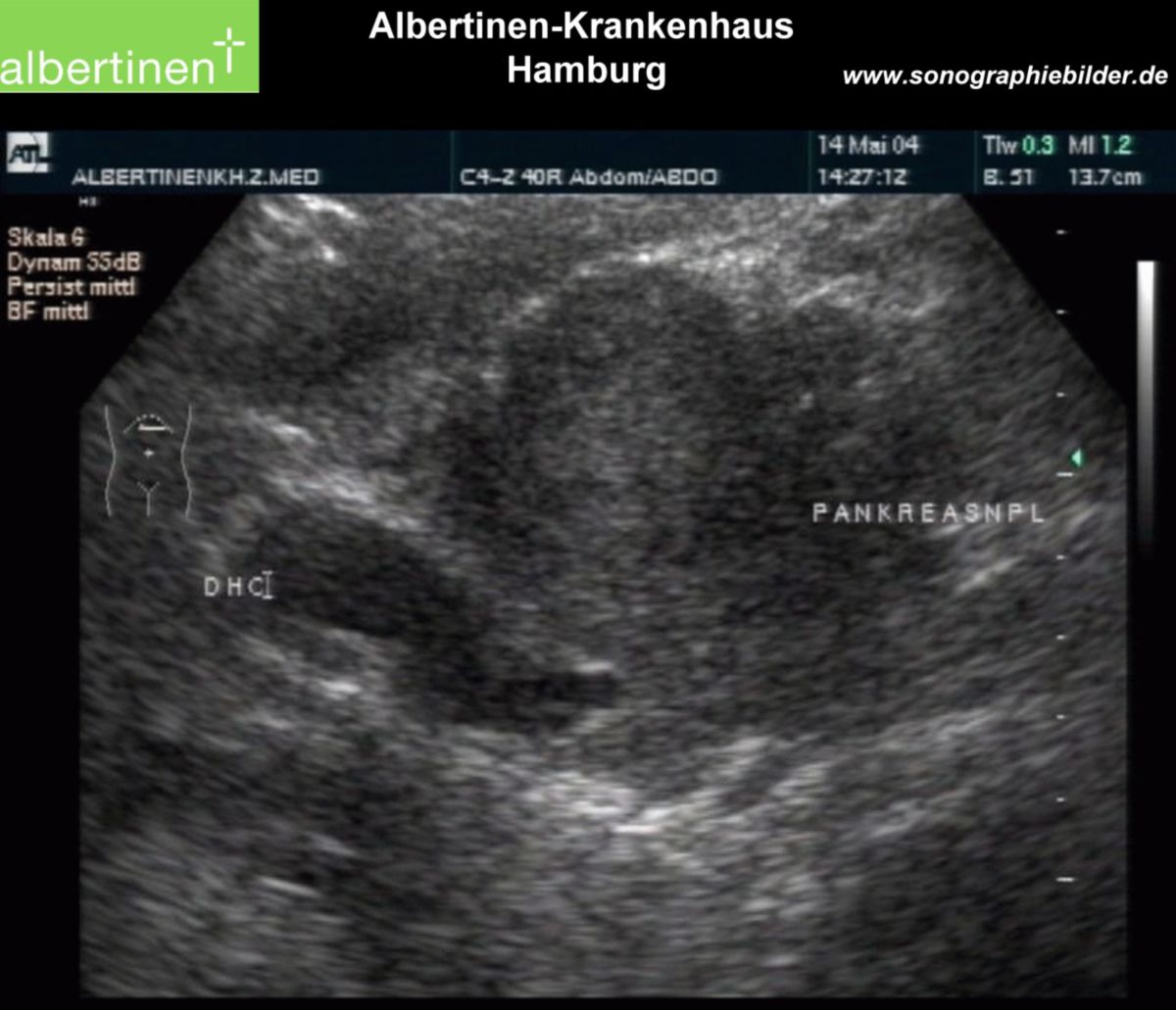
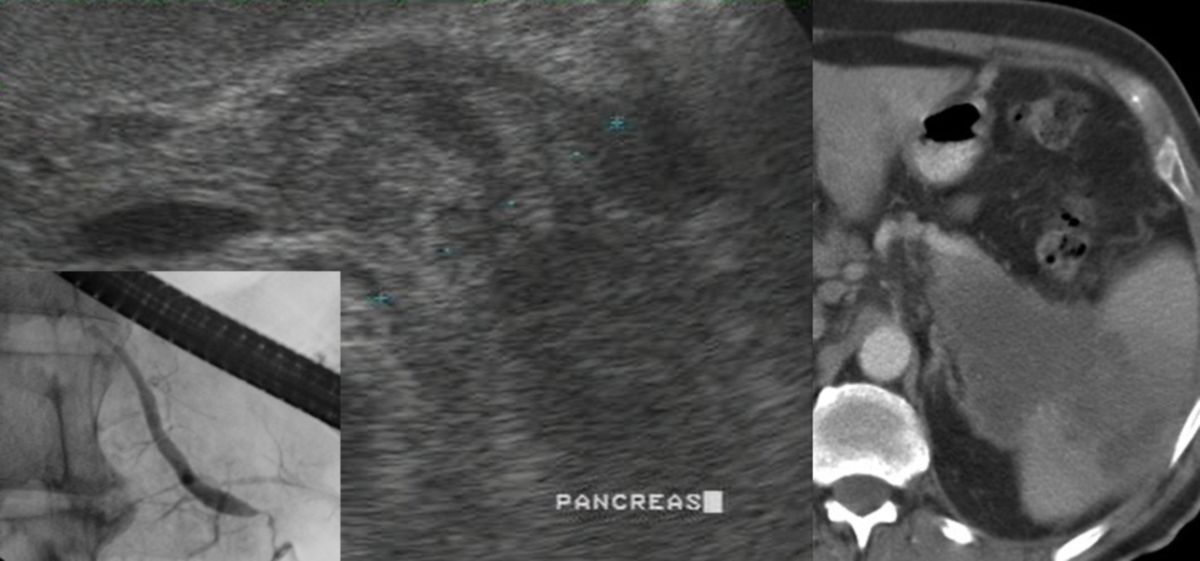
Bildgebung
Folgende bildgebende Verfahren werden in der Diagnostik des Pankreaskarzinoms eingesetzt:
- Sonographie als primäre Screeningmethode bei unklaren Oberbauchbeschwerden. Ziel ist die Darstellung des Pankreas (Form, Lage, Gangsystem). Insbesondere im Pankreaskopfbereich kann die Sonographie gut angewendet werden.
- Computertomographie (CT) und ggf. Magnetresonanztomographie (MRT) zur Darstellung des Karzinoms (insbesondere der Pankreasschwanzregion), zum Nachweis eines organüberschreitenden Wachstums bzw. einer Infiltration der Vena mesenterica inferior (Teardrop-Zeichen) und zur Metastasensuche. Entscheidend für die Beurteilung der Resektabilität.
- Endosonographie des Pankreas
- ERCP zur Darstellung der Gallen- und Pankreasgänge (Erweiterungen, Abbrüche, Stenosen), Möglichkeit zur Intervention mit Stenteinlage
CT-Fallbeispiel
Biopsie
Bei unsicheren Befunden kann z.B. unter sonographischer Kontrolle eine Biopsie durch Feinnadelpunktion gewonnen werden.
Labor
Bei klinischem oder bildmorphologischem Verdacht können Tumormarker bestimmt werden (CA 19-9 und CEA). Sie dienen zudem der Verlaufskontrolle. Eine Erhöhung kann auf einen Progress des Karzinoms hinweisen.
Laparoskopie
Bei resektablen Pankreaskarzinomen kann eine diagnostische Laparoskopie indiziert sein. Dies ist der Fall bei einer Tumorgröße > 3 cm, Aszites oder einem CA 19-9 > 500 U/ml (ohne Cholestase), da hier häufiger eine Mikrometastasierung vorliegt.
Weitere Diagnostik
Eine diagnostische Methode, die noch klinisch evaluiert wird, ist die Bestimmung von Mesothelin (MSLN), einem Glykoprotein der Zelloberfläche, das von Pankreaskarzinomzellen überexprimiert wird. Vor allem bei Patienten mit duktalen Adenokarzinomen des Pankreas ist das im Blut zirkulierende Mesothelin signifikant erhöht. Es eignet sich grundsätzlich auch zur Verlaufskontrolle von Patienten mit Pankreaskarzinomen. Als immunhistochemischer Marker ist Mesothelin bereits etabliert.
Therapie
Der einzig potenziell kurative Therapieansatz besteht in der chirurgischen R0-Resektion des Pankreaskarzinoms. Bei fehlender Resektabilität oder Fernmetastasierung liegt eine palliative Situation vor.
Chirurgische Therapie
Indikation
Entscheidend für die Resektabilität sind:
- Infiltration von Nachbarorganen: R0-Resektion grundsätzlich möglich
- Infiltration von Arterien: Bei Infiltration des Truncus coeliacus erfolgt nur in Einzelfällen eine Resektion.
- Infiltration von Venen: R0-Resektion bei Infiltration von Pfortader, Vena splenica und Vena mesenterica superior in Einzelfällen möglich.
Als grenzwertig resektable Tumoren gelten Pankreaskarzinome mit:
- Kontakt des Tumors zu Vena mesenterica superior und/oder Pfortader ohne Stenosierung der Gefäße durch Tumorkompression
- Ummauerung der Vena mesenterica superior und/oder der Pfortader ohne vollständige Ummauerung der benachbarten arteriellen Gefäße
- Kurzstreckiger Verschluss oder Stenose eines venösen Gefäßes durch einen Thrombus oder Tumoreinschluss, wenn proximal und distal des Verschlusses genug Gefäßmaterial für eine Rekonstruktion vorliegt
- Kurzstreckiger Einschluss oder Kontakt des Tumors mit der Arteria gastroduodenalis oder mit der Arteria hepatica propria, sofern der Truncus coeliacus nicht mit eingeschlossen ist
- Einschluss des Truncus coeliacus um < 180°
Verfahren
Ein kuratives Operationsverfahren beim Pankreaskopfkarzinom ist in der Regel die pyloruserhaltende partielle Duodenopankreatektomie nach Traverso-Longmire. Dabei wird neben dem Pankreaskopf ein Teil des Duodenums unter Erhalt des Pylorus sowie die Gallenblase und der Ductus choledochus entfernt. Weiterhin erfolgt eine Lymphadenektomie entlang des Ligamentum hepatoduodenale und entlang der Arteria hepatica propria bis zum Truncus coeliacus. Anschließend wird eine Rekonstruktion durch Pankreatikojejunostomie, Duodenojejunostomie und Anlage einer biliodigestiven Anastomose durchgeführt. Die Triangle-Operation, also die radikale Dissektion und Resektion im Dreieck zwischen Truncus coeliacus, Arteria mesenterica superior und der Vena portae, kann dabei möglicherweise das Risiko eines Lokalrezidivs senken.
Bei Infiltration des Bulbus duodeni wird die partielle Duodenopankreaktomie (Whipple-Operation) angewendet. Sie umfasst die Resektion von Pankreaskopf, distalem Magen, Duodenum, Gallenblase und Ductus choledochus sowie eine Lymphadenektomie und Rekonstruktion durch Roux-Y-Schlinge aus Jejunum und biliodigestiver Anastomose.
Karzinome des Pankreaskorpus und Pankreasschwanzes erfordern eine Pankreaslinksresektion mit Splenektomie oder eine totale Duodenopankreatektomie mit Splenektomie.
Nach (vollständiger) Entfernung des Pankreas sind sowohl die exokrin als auch die endokrin sezernierten Enzyme des Pankreas zu substituieren.
Bei Diagnosestellung ist bei nur 15 bis 20 % der Patienten eine kurative Operabilität gegeben. Die operative Letalität liegt in Schwerpunktzentren bei unter 5 %. Nach R0-Resektion liegt das mediane Überleben bei ca. 40 Monaten und das 5-Jahres-Überleben bei ca. 40 %. Nach R1-Resektion liegt das mediane Überleben bei unter 23 bis 27 Monaten. Bei Pankreaslinksresektion liegt das mediane Überleben nach R0-Resektion bei 60 Monaten und das 5-Jahres-Überleben bei ca. 50 %.
Chemotherapie bei kurativem Therapiekonzept
Adjuvante Chemotherapie
Bei resektablen PDAC erfolgt nach kurativ intendierter Resektion eine adjuvante Chemotherapie. Bei gutem Allgemeinzustand wird das modifizierte, d.h. dosisreduzierte FOLFIRINOX-Regime (mFOLFIRINOX) empfohlen:
Bei eingeschränktem Allgemeinzustand können Gemcitabin mit/ohne Capecitabin eingesetzt werden.
Mit adjuvanter Chemotherapie mit mFOLFIRINOX liegt das mediane Überleben bei ca. 54 Monaten, unter Gemicitabin bei 35 Monaten.
Neoadjuvante Chemotherapie
Bei grenzwertig resektablem oder lokal fortgeschrittenem, nicht resektablem PDAC erfolgt eine neoadjuvante Chemotherapie mit FOLFIRINOX. Das 12-Monats-Überleben liegt bei grenzwertig resektablem Tumor und anschließender OP bei etwa 84 %. Bei lokal fortgeschrittenem, nicht resektablem PDAC kann nach neoadjuvanter FOLFIRINOX-Gabe eine Resektionsrate von rund 60 % erzielt werden. Das mediane Gesamtüberleben liegt nach FOLFIRINOX und Resektion bei 16 Monaten und die 3-Jahres-Überlebensrate bei 28 %.
Palliative Chemotherapie
Möglichkeiten der palliativen Chemotherapieregime sind z.B. FOLFIRINOX oder Gemcitabin bzw. Gemcitabin in Kombination mit nab-Paclitaxel oder Erlotinib. Auf Basis neuer Studienergebnisse wird zur Erstlinientherapie zudem das Kombinationsregime NALIRIFOX empfohlen. Stand 2024 handelt es sich um einen Off-Label-Use.
Bei der Auswahl der passenden Therapie spielen u.a. der ECOG-Performance Status, Komorbiditäten und die Präferenz der Betroffenen eine wichtige Rolle.
Zielgerichtete Therapie
Ungefähr 4 bis 7 % der Patienten mit PDAC weisen eine BRCA1/2-Keimbahnmutation auf. Bei metastasiertem PDAC und Nachweis dieser Mutation wird eine Platin-basierte Erstlinientherapie bevorzugt. Falls nach mindestens 16 Wochen kein Tumorprogress vorliegt, kann eine Erhaltungstherapie mit Olaparib zum Einsatz kommen. Das mediane progressionsfreie Überleben liegt dann bei 7,4 Monaten (vs. 3,8 Monaten unter Placebo).
Über 90 % der Patienten mit PDAC zeigen eine onkogene KRAS-Mutation. Bei dem seltenen Vorliegen einer KRAS-G12C-Mutation kann nach Ausschöpfen aller Therapieoptionen ein selektiver KRAS-G12C-Inhibitor zum Einsatz kommen. Weitere molekulare Veränderungen mit therapeutischer Relevanz sind BRAF-Mutationen und Fusionen in verschiedenen Genen wie NTRK, ROS1, ALK, RET und NRG1.
Immuntherapie
Bei 1 % der PDAC sind Mikrosatelliteninstabilitäten zu finden. Dann können in bestimmten Fällen Checkpoint-Inhibitoren in Frage kommen.
Supportive Therapien
In allen Phasen der Erkrankungen sollten supportive Therapien angeboten werden. Diese sind durch standardisierte Screenings auf typische beeinträchtigende Symptome und psychosoziale Belastungen zu ergänzen.
Nach operativer Therapie sollte eine Anschlussheilbehandlung und eine strukturierte Nachsorge angeboten werden. In palliativer Situation sollten Betroffene, unabhängig vom Krankheitsstadium, früh Informationen und Angebote hinsichtlich einer Palliativversorgung erhalten. Der Bedarf sollte durch spezialisierte Teams ermittelt und die Versorgung an die Komplexität der jeweiligen Situation angepasst werden.
Um eine möglichst hohe Lebensqualität der Betroffenen zu erhalten, ist eine effektive Schmerztherapie notwendig. Hier steht die medikamentöse Analgesie oder die Plexus-coeliacus-Blockade zur Verfügung. Auch eine Schmerzanamnese mit schmerzbezogener klinischer Untersuchung sollte regelmäßig erfolgen.
Mittels etablierter Verfahren sollte zudem regelmäßig auf Hinweise einer Mangelernährung hin untersucht werden. Eine qualifizierte Fachkraft für Ernährung ist bei Problemen hinzuzuziehen und u.U. eine enterale oder parenterale Ernährung einzuleiten. Insbesondere bei palliativer Behandlung bzw. bei fortgeschrittenen Befunden sind unter anderem auch die Erhaltung der Durchgängigkeit des Gastrointestinaltraktes und die Behebung einer Cholestase wichtige Therapieziele (maligne Obstruktion). Dies kann durch Einlage von Stents oder palliative Operationen zur Stabilisierung des Galledurchflusses (biliodigestive Anastomose) erfolgen.
Bei an einem fortgeschrittenen Pankreaskarzinom erkrankten Personen in ambulanter Behandlung kann weiterhin die Gabe von niedermolekularem Heparin als antikoagulatorische Primärprophylaxe unter Nutzen-Risiko-Abwägung erfolgen.
Prävention
Ein Screening auf das Vorliegen eines Pankreaskarzinoms kann bei bestimmten Personengruppen sinnvoll sein, z.B. bei Erfüllung der Kriterien für ein familiäres Pankreaskarzinom oder Vorliegen einer hereditären chronischen Pankreatitis. Mögliche empfohlene Verfahren sind eine MRT/MRCP sowie ein endoskopischer Ultraschall. Die Zeitpunkte, an denen mit einem Screening begonnen werden sollte, unterscheiden sich je nach Risikokonstellation. Asymptomatischen Personen ohne erhöhtes Risiko oder mit erhöhtem Risiko für ein sporadisches Pankreaskarzinom wird kein Screening empfohlen.
Prognose
Die Gesamt 5-Jahres-Überlebensrate liegt aktuell (2023) bei 10 %.[2] Die mittlere Überlebensrate bei palliativen Maßnahmen beträgt 6 bis 9 Monate.
Details zur Prognose in Bezug auf unterschiedliche Therapieprinzipien sind unter dem Abschnitt "Therapie" aufgelistet.
Leitlinie
- AWMF - S3-Leitlinie Exokrines Pankreaskarzinom, Stand 31.03.2024
Quellen
- ↑ Tsai H-J et al.: Environmental risk factors of pancreatic cancer. J Clin Med 2019; 8: 1427
- ↑ Zantrum für Krebsregisterdaten – Bauchspeicheldrüsenkrebs (Pankreaskarzinom), abgerufen am 10.05.2023
Literatur
- Onkopedia - Pankreaskarzinom, abgerufen am 10.05.2023