Akute myeloische Leukämie











Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenEnglisch: acute myeloid leukemia
Definition
Die akute myeloische Leukämie, kurz AML, ist eine biologisch heterogene, maligne Erkrankung des blutbildenden Systems. Sie geht von entarteten myeloischen Progenitorzellen oder Stammzellen aus und tritt überwiegend bei Erwachsenen über 60 Jahren auf. Ohne adäquate Therapie verläuft sie meist rasch progredient mit ungünstiger Prognose.
Epidemiologie
Die AML ist eine Leukämie des höheren Erwachsenenalters; das mediane Erkrankungsalter liegt bei etwa 68 Jahren. Die Inzidenz beträgt in der Gesamtbevölkerung etwa 4,3 Erkrankungen pro 100 000 Einwohner und Jahr. In der Altersgruppe über 65 Jahre steigt sie auf über 20 Fälle pro 100 000 Einwohner an.[1][2]
Ätiologie
Die akute myeloische Leukämie wird durch eine Reihe sehr unterschiedlicher zytogenetischer Veränderungen ausgelöst, die auf der Ebene des Genoms (Monosomien, Trisomien), der Chromosomen (Translokationen, Inversionen) und/oder der Gene (Genmutationen) auftreten können. Meist betreffen diese Veränderungen den hochproliferativen Progenitorpool, d.h. CD34+/CD38+-Zellen, seltener den Stammzellpool (CD34+/CD38--Zellen).[3]
Die genauen Ursachen sind nicht vollständig bekannt. Es werden jedoch Umweltfaktoren, genetische Prädispositionen und hämatologische Vorerkrankungen als entscheidende Risikofaktoren angenommen.
Umweltfaktoren und exogene Noxen
Die AML tritt gehäuft nach Exposition gegenüber bestimmten kanzerogenen Substanzen auf:
- Benzol und andere Bestandteile von Mineralölprodukten
- Pflanzenschutzmittel wie Herbizide und Pestizide
- Ionisierende Strahlung
- Zigarettenrauchen
- Chemotherapeutika, insbesondere Alkylanzien und Topoisomerase-II-Inhibitoren
Diese Substanzen führen u.a. zu epigenetischen Veränderungen und DNA-Schäden, welche die Leukämieentstehung fördern.[4]
Genetische Prädisposition
Ein erhöhtes Risiko besteht bei bestimmten hereditären Erkrankungen:
- Trisomie 21 (etwa 20-fach erhöhtes Risiko)
- Fanconi-Anämie
- Bloom-Syndrom, Schwachmann-Diamond-Syndrom, Neurofibromatose Typ 1 (seltener)[5]
Zunehmend erkannt wird die sogenannte klonale Hämatopoese unbestimmten Potenzials (CHIP). Dabei kommt es – vor allem im Alter – zur Akkumulation somatischer Mutationen in hämatopoetischen Zellen ohne Leukämiezeichen. Liegt gleichzeitig eine Zytopenie vor, spricht man von einer klonalen Zytopenie unklarer Signifikanz (CCUS). Beide gelten als mögliche Vorläuferzustände der AML.
Sekundäre AML
Die AML kann sich sekundär aus anderen hämatologischen Erkrankungen entwickeln, z.B.:
- aus einer Myelodysplasie
- aus einer myeloproliferativen Neoplasie, z.B. Osteomyelofibrose oder Chronische myeloische Leukämie
- nach Immunsuppression, Bestrahlung oder Organtransplantation
Im letzteren Fall spricht man von einer therapieassoziierten AML (t-AML).[6]
Mutationen
Es gibt eine Vielzahl verschiedener Mutationen, die auf zellulärer Ebene für die AML verantwortlich sind. Sie unterscheiden sich nicht nur interindividuell, sondern auch intraindividuell, d.h. selbst bei einem Patienten können verschiedene Mutationen ("Subklone") festgestellt werden, deren Zusammensetzung sich im Laufe der Erkrankung auch ändern kann.
Beispiele für typische AML-Mutationen sind die interne Tandemduplikation FLT3-ITD und die t(15;17)-Translokation. Am häufigsten finden sich bei der AML Mutationen in den Genen FLT3, NPM1 und DNMT3A. Dabei ist besonders die FLT3-ITD-Mutation prognostisch ungünstig. NPM1-Mutationen bilden laut WHO-Klassifikation von 2022 eine eigenständige AML-Entität, auch bei einem Blastenanteil unter 20 %.
Weitere relevante Veränderungen finden sich in den sogenannten WIT-Genen: WT1, IDH1, IDH2 und TET2. CEBPA-Mutationen treten vor allem in biallelischer ("double-hit") Form auf und definieren eine eigenständige prognostisch günstige AML-Entität.[7]
Morphologie
Die akute myeloische Leukämie ist durch einen sogenannten Differenzierungsblock auf der Ebene der myeloischen multipotenten Progenitorzellen gekennzeichnet. Das Knochenmark gibt die unreifen Myeloblasten aufgrund mangelnder Reifungskapazität vermehrt ins Blut ab. Im Blut finden sich Vorstufen der Granulozytopoese wie Promyelozyten, Myeloblasten und Monozyten. Diese imponieren meist als Zellen mit großem Zellkern, schmalem Zytoplasma und häufig vorkommenden Auerstäbchen(Myeloperoxidase-positive Einschlusskörper).
Im Knochenmark kommt es zu einer Verdrängung der Erythropoese und Megakaryopoese, was zu einer Depletion von Granulozyten, Thrombozyten und Erythrozyten führt. Die leukämischen Infiltrate imponieren makroskopisch als grau-rote Areale.
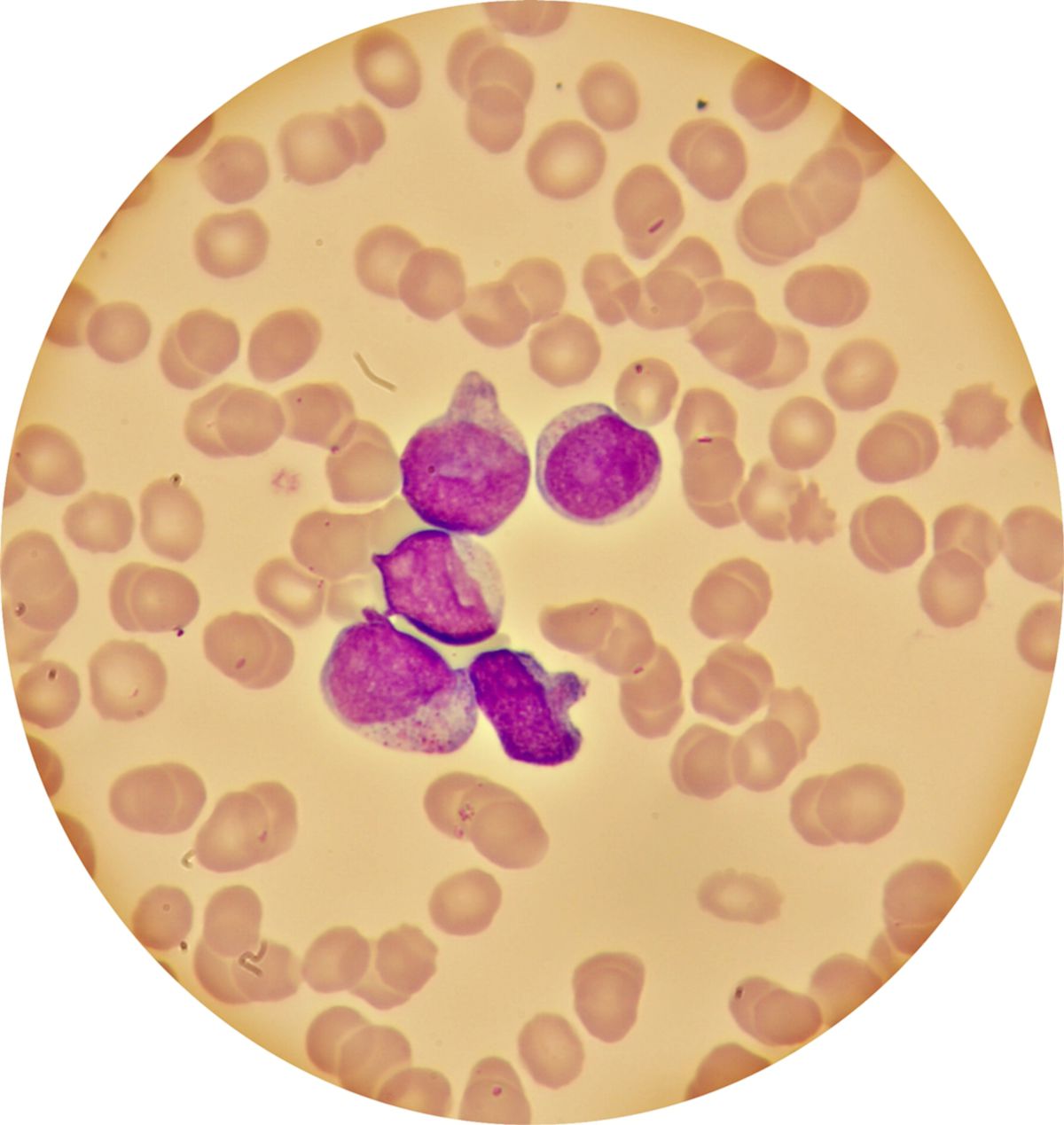
Klinik
Bei der AML handelt es sich um ein akutes Krankheitsbild, weshalb sich Allgemeinsymptome (Schwäche, Fieber, Nachtschweiß) typischerweise mit kurzer Anamnese zeigen. Da sich die leukämischen Zellen in Knochenmark und Blut ausbreiten, entwickeln sich Symptome einer gestörten Hämatopoese:[8]
- Infektanfälligkeit durch Fehlen reifer und funktionsfähiger Leukozyten (z.B. Mundsoor)
- Gerinnungsstörungen (Petechien, Epistaxis, Hämatome)
- Anämie mit Blässe, Schwäche und Dyspnoe
Weitere organspezifische Symptome sind Splenomegalie, Hepatomegalie und selten auch Lymphknotenschwellungen.
Einige Unterformen der AML können spezifische Symptome hervorrufen. So kann bei einer Schleimhautinfiltration durch Blasten bei der myelomonozytären (M4) und monozytären (M5) Leukämie eine Gingivahyperplasie auffallen.
Diagnostik
Die Basisdiagnostik der AML umfasst neben Anamnese und körperlicher Untersuchung (Zeichen der gestörten Hämatopoese, Organvergrößerungen) eine Blutuntersuchung einschließlich Differentialblutbild. Typische Befunde sind hierbei:
- Anämie, Thrombozytopenie und Granulozytopenie. Die Leukozytenzahl ist kein zuverlässiger Marker einer akuten Leukämie. Je nachdem, ob ein leukämischer oder aleukämischer Verlauf vorliegt, können erhöhte, normale und auch erniedrigte Leukozytenzahlen im peripheren Blut vorkommen.
- leukämische Blasten, fehlende Zwischenstufen (Hiatus leucaemicus)
- BSG erhöht
- Harnsäure und LDH erhöht (aufgrund des vermehrten Zellumsatzes)
Zur Sicherung der Diagnose wird eine Knochenmarkpunktion durchgeführt; als generelle Schwelle gelten mehr als 20 % Blastenzellen im Knochenmark oder im peripheren Blut, wobei bei bestimmten genetisch definierten Subtypen auf diese Schwelle verzichtet wird.
Zur Klassifikation sind weitere Untersuchungen notwendig:
Ist der Patient für eine Stammzelltransplantation vorgesehen, erfolgt zusätzlich die HLA-Typisierung und die Überprüfung des CMV-Status
Ergänzende Diagnostik
Einteilung
WHO-Klassifikation
Die aktuell (2025) gültige Einteilung der AML folgt der WHO-Klassifikation von 2022. Diese richtet sich nach molekular- und zytogenetischen Kriterien und unterteilt in die folgenden Subgruppen:[9]
- AML mit genetisch definierenden zytogenetischen Aberrationen (Translokationen, Inversionen, Mutationen)
- AML mit t(8;21)(q22;q22); RUNX1-RUNX1T1
- AML mit inv(16)(p13.1q22) oder t(16;16)(p13.1;q22); CBFB-MYH11
- akute Promyelozytenleukämie (APL) mit t(15;17)(q22;q12); PML-RARA
- AML mit t(9;11)(p22;q23); KMT2A-MLLT3
- AML mit t(6;9)(p23;q34); DEK-NUP214
- AML mit inv(3)(q21q26.2) oder t(3;3)(q21;q26.2); GATA2, MECOM
- AML (megakaryoblastär) mit t(1;22)(p13;q13); RBM15-MKL1
- vorläufige Entität: AML mit BCR-ABL1
- AML mit mutiertem NPM1
- AML mit biallel mutiertem CEBPA
- AML, myelodysplasia-related (AML-MR)
- therapieassoziierte myeloide Neoplasien (tAML)
- nicht anderweitig spezifizierte AML (not otherwise specified, NOS)
- myeloisches Sarkom (extramedullärer myeloischer Tumor, Chlorom)
- myeloische Proliferationen bei Down-Syndrom
- akute Leukämien mit unklarer Linie
Risikogruppen
Laut den Kriterien des European LeukemiaNet (ELN) 2022 werden die akuten myeloischen Leukämien in drei prognostische Gruppen eingeteilt:
- Günstig: z. B. t(8;21)(q22;q22); RUNX1‑RUNX1T1
- Intermediär: z.B. t(9;11)(p22;q23)
- Ungünstig: z.B. t(6;9)(p23;q34)
FAB-Klassifikation
Die Einteilung erfolgt nach den Vorschlägen der French-American-British Cooperative-Group (FAB, FAB-Klassifikation) und richtet sich nach zytomorphologischen Kriterien.
- M0: AML mit minimaler Differenzierung
- M1: AML ohne Ausreifung (ca. 20 %)
- M2: AML mit Ausreifung (ca. 30 %)
- M3: Promyelozytenleukämie (ca. 5 %)
- M4: Myelomonozytäre Leukämie (ca. 30 %)
- M5: Monozytäre Leukämie (ca. 10 %)
- M6: Erythroleukämie (selten)
- M7: Megakariozytenleukämie (selten)
Eine detailliertere Darstellung mit Zytochemie und Abberationen findet sich auf der eigenen Seite der FAB-Klassifikation.
Therapie
Grundsätzlich besteht die kurative Therapie der AML aus einer intensivierten Chemotherapie, die in mehrere Phasen unterteilt wird.
- In der Induktionsphase wird mit hochdosierten Chemotherapeutika (z. B. Daunorubicin, Cytosin-Arabinosid) über mehrere Wochen eine massive Reduktion der Tumorzellen und das Erreichen einer Vollremission angestrebt.
- Als Konsolidierungsphase bezeichnet man die darauffolgende zytostatische Therapie mit mittelhoch dosierten Therapeutika über mehrere Monate, oftmals gefolgt von einer allogenen Stammzelltransplantation bei geeigneten Patienten.[10]
- Anschließend folgt eine Erhaltungstherapie über bis zu zwei Jahre mit niedriger Dosierung. Eine generelle Erhaltungstherapie ist jedoch nicht mehr Standard, sondern wird gezielt bei bestimmten Subtypen eingesetzt.
Ist ein sofortiger Therapiebeginn notwendig, z.B. aufgrund eines lebensbedrohlichen Leukostasesyndroms, werden die Ergebnisse der molekulargenetischen Untersuchungen nicht abgewartet und direkt eine Induktionstherapie nach dem sogenannten Standardschema eingeleitet - auch "7+3-Schema" genannt. Dieses besteht aus der Gabe von Cytarabin an den Tagen 1 bis 7 mit einem Anthracyclin (z.B. Daunorubicin) an den Tagen 1 bis 3. Wird im Verlauf eine FLT3-Mutation nachgewiesen, erfolgt die zusätzliche Gabe des Multikinase-Inhibitors Midostaurin.
Eine weitere Therapieoption mit kurativem Ansatz ist die allogene Stammzelltransplantation. Sie wird empfohlen bei:
- Hochrisiko-AML mit ungünstiger Zytogenetik
- therapieassoziierter AML oder sekundärer AML
- FLT3-Mutation mit persistierender MRD nach Induktionstherapie
Im Anschluss an die allogene Stammzelltransplantation kann bei FLT3-ITD-positiven Patienten eine Erhaltungstherapie mit einem Tyrosinkinaseinhibitor wie Midostaurin, Gilteritinib oder Sorafenib erfolgen.
Eine potentiell kurative Therapie kommt in der Regel nur für Patienten unter 75 Jahren mit wenigen Komorbiditäten in Betracht. Andernfalls wird eine palliative Therapie verfolgt, z. B. mit:
- Hypomethylierende Substanzen (HMA) wie Azacitidin oder Decitabin in Kombination mit Venetoclax
- Low-Dose-Cytarabin (LDAC)
- Best Supportive Care
Zur Behandlung der Promyelozytenleukämie (M3) werden auch Vitamin-A-Präparate wie ATRA in Kombination mit Arsentrioxid eingesetzt.
Prognose
Nach erfolgreicher Induktionstherapie erreichen jüngere und therapiefähige Patienten häufig eine Vollremission – das heißt eine vollständige Normalisierung des Blutbilds und das Fehlen extramedullärer Manifestationen.
Trotz Remission versterben viele Patienten im Verlauf durch einen Rückfall oder Therapiekomplikationen; die 5-Jahres-Überlebensrate liegt in aktuellen Real-World-Daten bei etwa 18 bis 20 %.[11]
Prognostisch ungünstige Faktoren für den Verlauf und die Therapie sind unter anderem:
- Alter > 60 Jahre
- Initiale Leukozytenzahl > 100 000/µl
- Komplexe chromosomale Aberrationen
Zusätzlich zunehmend relevant sind Genmutationen wie TP53, ASXL1 oder eine hohe MRD-Last nach Induktion.
Quellen
- ↑ SEER Cancer Stat Facts – AML, abgerufen am 20.11.2025
- ↑ Shallis et al., Epidemiology of acute myeloid leukemia: Recent progress and enduring challenges, Blood Rev, 2019
- ↑ Shlush, Age-related clonal hematopoiesis, Blood, 2018
- ↑ Khalade et al., Exposure to benzene at work and the risk of leukemia: a systematic review and meta-analysis, Environ Health, 2010
- ↑ Mangaonkar und Patnaik, Hereditary Predisposition to Hematopoietic Neoplasms: When Bloodline Matters for Blood Cancers, Mayo Clin Proc, 2020
- ↑ Döhner et al., Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN, Blood, 2022
- ↑ Nong et al., Common Driver Mutations in AML: Biological Impact, Clinical Considerations, and Treatment Strategies, Cells, 2024
- ↑ Shimony et al., Acute Myeloid Leukemia: 2025 Update on Diagnosis, Risk-Stratification, and Management, Am J Hematol, 2025
- ↑ Khoury et al., The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms, Leukemia, 2022
- ↑ Magee und Grunwald, Updates on Therapy Options in Fit and Unfit Patients with Newly Diagnosed AML, Curr Treat Options Oncol, 2025
- ↑ Liu et al., 1- and 5-Year Survival for Adults with Acute Myeloid Leukaemia and 30-Day Mortality after Initial Systemic Anti-Cancer Therapy, with an Exploration of Factors Associated with Poorer Outcomes: Data from a National Registry in England, 2013-2020, Blood, 2023