Morbus Fabry










Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenSynonyme: Fabry-Krankheit, Fabry-Syndrom oder Fabry-Anderson-Krankheit, Alpha-Galaktosidase-A-Mangel, diffuses Angiokeratom, Angiokeratoma corporis diffusum Fabry
Englisch: Fabry disease, Anderson-Fabry disease, alpha-galactosidase A deficiency
Definition
Der Morbus Fabry ist eine X-chromosomal vererbte, lysosomale Speicherkrankheit. Ein Defekt des Enzyms alpha-Galaktosidase A (GLA) führt zu einer intrazellulären Akkumulation von Sphingolipiden in verschiedenen Organen.
Epidemiologie
Die Prävalenz beträgt 1:40.000 bis 1:117.000 Lebendgeburten. Es wird jedoch von einer hohen Dunkelziffer ausgegangen. In einigen Screeningprogrammen wird eine Inzidenz von 1/4.600 bis 1/1.250 beschrieben.[1][2]
Das mittlere Manifestationsalter liegt bei Männern mit klassischer genetischer Variante zwischen 3 und 10 Jahren, bei Frauen zwischen 6 und 15 Jahren. Während Männer häufig früher und schwerer erkranken, können auch Frauen mit Morbus Fabry jede Krankheitsschwere erreichen.
Ätiopathogenese
Die Erkrankung beruht auf X-chromosomal-rezessiv vererbten Defekten des lysosomalen Enzyms alpha-Galaktosidase A. Über 900 verschiedene Mutationen des hierfür kodierenden GLA-Gens sind beschrieben. Dabei handelt es sich meist um Nonsense- oder Missense-Varianten. Der Enzymdefekt führt zur gestörten Metabolisierung und intrazellulären Akkumulation von Glykosphingolipiden wie Globotriaosylceramid (Gb3) und seiner deacetylierten Fom (Lyso-Gb3). Gb3 beeinflusst Prozesse der Inflammation, ischämischen Hypertrophie und Fibrose. Lyso-Gb3 fördert die Proliferation der glatten Muskulatur.
Die Ablagerungen finden sich unter anderem in:
- Nieren: Podozyten, Mesangialzellen, Endothelzellen, Tubulusepithelzellen
- Herz: Myokardzellen, Endothelzellen, Fibroblasten, spezifisches Reizleitungssystem
- PNS: Neurone der Spinalganglien und des autonomen Nervensystems
- ZNS: Neurone
- Blutgefäße: Endothelzellen, glatte Muskelzellen
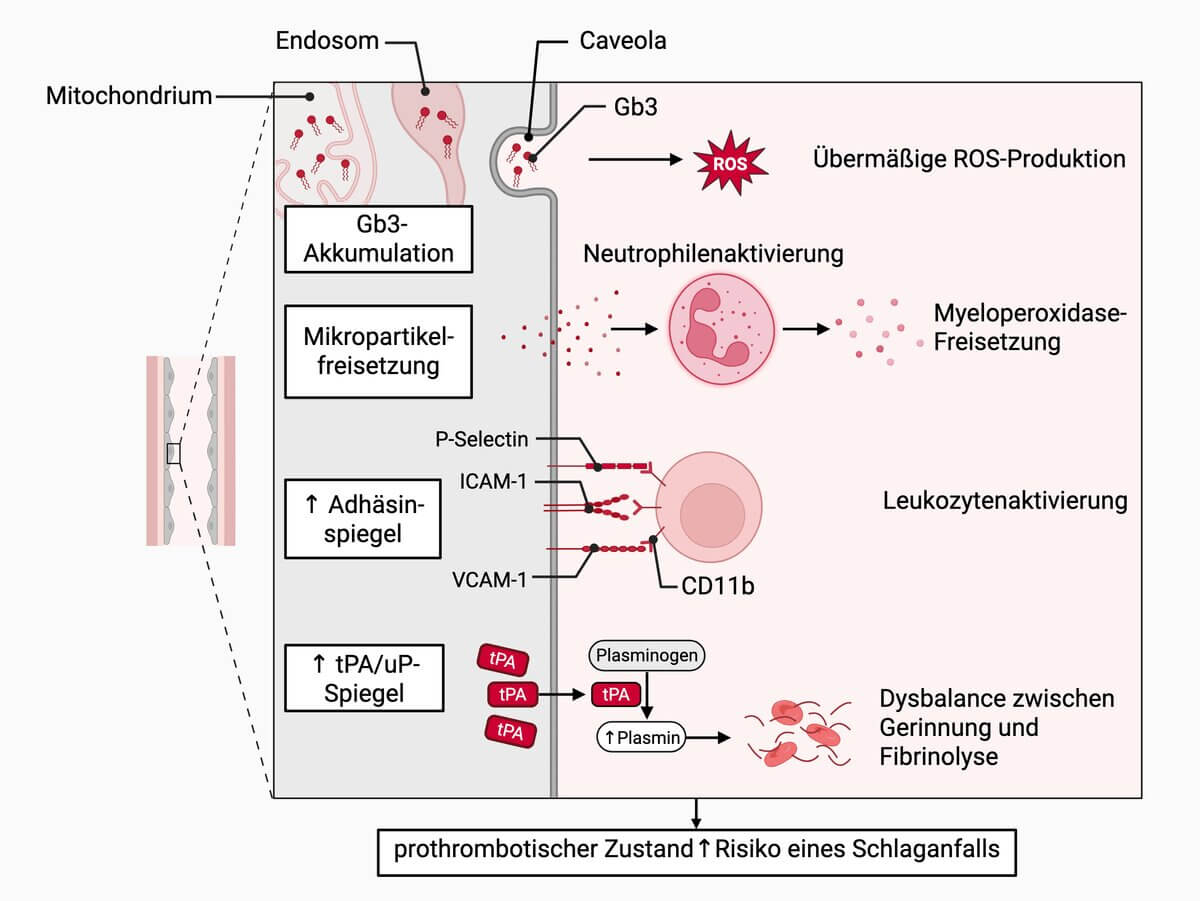
Klinik
Der klinische Verlauf ist interindividuell sehr verschieden und kann nicht vorausgesagt werden. Die Zahl und Schwere der Symptome nehmen mit dem Alter zu. Die Erkrankung beginnt in der Regel im Kindes- und Jugendalter mit folgenden Symptomen:
- Niere: Mikroalbuminurie, Proteinurie
- Herz: autonome kardiale Fehlregulation, z.B. mit abnormer Herzfrequenzvariabilität
- Fabry-assoziierte Schmerzen an Händen und Füßen
- Hitze- und/oder Kälteintoleranz
- Hypohidrose
- Dreh- und Schwankschwindel (akut und permanent)
- wiederhole Hörstürze, Tinnitus
- gastrointestinale Beschwerden, Bauchschmerzen
- Fatigue
- Augenveränderungen, z.B. Cornea verticillata
- Angiokeratome
Im frühen Erwachsenenaler (17. bis 30. Lebensjahr) kommen weitere Beschwerden dazu:
- Niere: Proteinurie, fortschreitende Niereninsuffizienz
- Herz: hypertrophe Kardiomyopathie (HCM), pathologische Bradykardien, chronotrope Inkompetenz, Arrhythmien
- TIA, Schlaganfall
- kognitive und affektive Symptome (z.B. Depression)
Im späten Erwachsenenalter kann es zu einer Progression bis hin zu Niereninsuffizienz, Herzinsuffizienz und rezidivierenden Schlaganfällen kommen.
Renal
Patienten mit Mikroalbuminurie unklarer Genese oder Proteinurie sowie eingeschränkter Nierenfunktion unklarer Genese sollten auf das Vorliegen eines Morbus Fabry untersucht werden. Im Mittel werden erste klinische Zeichen der Nephropathie im Alter von 20 Jahren beobachtet, während eine terminale Niereninsuffizienz meist mit 38 Jahren eintritt.
Der elektronenmikroskopische Nachweis von Gb3-Ablagerungen in einer Nierenbiopsie kann durchgeführt werden, wenn die übliche Diagnostik nicht eindeutig ist. Die histologische Diagnose der Fabry-Nephropathie erfolgt lichtmikroskopisch (Toluidinblau-Färbung). Immunhistologisch oder elektronenmikroskopisch zeigen sich meist zwiebelschalenförmige Ceramidablagerungen ("Zebra-Körperchen"). Im Verlauf kommt es zu einer progredienten Nierenparenchymschädigung mit interstitieller Fibrose und fokaler Glomerulosklerose.
Häufig finden sich multiple, meist uniform kleine, subkapsuläre Nierenzysten.
Kardial
Über 50 % aller Fabry-Patienten weisen bereits im Alter von 36 Jahren eine kardiale Beteiligung auf. Mehr als die Hälfte entwickeln eine Kardiomyopathie. Typische Befunde sind:
- Anfangs linksventrikuläre, meist konzentrische Hypertrophie. Im Spätstadium asymmetrische Hypertrophie mit verdicktem Kammerseptum und Ausdünnung der posterolateralen Wand. Die ausgedünnten Areale zeigen regionale Wandbewegungsstörungen. Nur selten zeigt sich eine Obstruktion des linksventrikulären Ausflusstrakts (LVOT) im Sinne einer hypertrophen obstruktiven Kardiomyopathie (HOCM).
- Intramyokardiale Fibrose: Late Enhancement in der Kardio-MRT. Indirekt kann die Fibrose auch in der Echokardiographie mittels "speckle tracking imaging" dargestellt werden. Bei Frauen kann auch eine intramyokardiale Fibrose ohne LVH vorliegen.
- EKG-Veränderungen wie kurze P-Welle, kurzes PR-Intervall, Zunahme der QRS-Zeit und Repolarisationsstörungen
- Herzrhythmusstörungen, z.B. paroxysmales oder permanentes Vorhofflimmern, nicht anhaltende oder anhaltende ventrikuläre Tachykardien
- Klappenvitien (Mitral- und Aortenklappe)
Myokardinfarkt, Herzinsuffizienz und plötzlicher Herztod durch maligne Arrhythmien zählen zu den Haupttodesursachen. Die fortgeschrittene Kardiomyopathie kann zu Vorhofflimmern und somit zu embolischen Schlaganfällen führen.
Neurologisch
PNS
Fabry-assoziierte Schmerzen kommen bei 33 bis 80 % der Männer und 25 bis 70 % der Frauen vor. Am häufigsten treten sie episodisch in Form von auslösbaren Schmerzattacken, Allodynie oder Hyperalgesie auf. Als Schmerzkrisen weden sehr starke Schmerzen bezeichnet, die sich über den ganzen Körper ausdehnen und Stunden bis Tage anhalten können. Die Schmerzen treten meist an den Akren auf und sind mit Brennen, Kribbeln und Taubheitsgefühl verbunden. Grundsätzlich kann jede Körperregion (inkl. Kopfschmerzen, Gelenkschmerzen) betroffen sein. Typische Auslöser sind Hitze, Kälte, körperliche Aktivität und Fieber.
Bei Morbus Fabry ist die Small-Fiber-Neuropathie (SFN) deutlich häufiger als eine Polyneuropathie. Die SFN trägt wahrscheinlich zu den Fabry-assoziierten Schmerzen bei. Bei Verdacht auf eine SFN sollte eine Hautstanzbiopsie vom lateralen Unter- und Oberschenkel oder Rücken zur Bestimmung der intraepidermalen Nervenfaserdichte und eine quantitative sensorische Testung zur Bestimmung der thermischen Wahrnehmungs- und Schmerzschwellen erfolgen. In der Biopsie zeigt sich ein Denervierungsmuster. Bei der QST finden sich angehobene Kaltdetektionsschwellen.
Weiterhin treten Engpasssyndrome (z.B. Karpaltunnelsyndrom) besonders im jungen Alter gehäuft auf.
ZNS
Fast 25 % der Patienten entwickeln im Verlauf der Erkrankung ein zerebrovaskuläres Ereignis (TIA, Schlaganfall). Das mittlere Alter beträgt 34 Jahre (Männer) bzw. 54 Jahre (Frauen). Ein Schlaganfall kann auch die Erstmanifestation darstellen. Insbesondere bei jungen Patienten mit zerebraler Ischämie unklarer Genese muss an einen Morbus Fabry gedacht werden.
Eine häufige zerebrale Manifestation des Morbus Fabry ist eine Vaskulopathie der kleinen Gefäße (Mikroangiopathie), deren Pathophysiologie unklar ist. Möglicherweise spielen klassische zerebrovaskuläre Risikofaktoren eine Rolle. In der MRT finden sich entsprechend Marklagerläsionen ("white matter lesions"). Die klinisch meist asymptomatische Leukenzephalopathie zeigt meist eine symmetrische Verteilung ohne spezifisches Muster. Ausgeprägte Befunde mit konfluierenden und großflächigen Marklagerläsionen sind eher selten.
In der MRT treten außerdem T1w-Signalanhebungen in den Basalganglien, insbesondere im Pulvinar thalami auf.
Neuropsychiatrisch
Depressive Symptome sind bei Patienten mit Morbus Fabry häufig. Weiterhin werden kognitive Symptome wie Konzentrationsstörungen oder Vergesslichkeit beschrieben.
Gastrointestinal
Ungefähr 50 bis 70 % der Fabry-Patienten weisen gastrointestinale Beschwerden auf, insbesondere im jüngeren Lebensalter. Beschrieben werden Völlegefühl, Bauchschmerzen, Diarrhö, Obstipation, Übelkeit und Erbrechen. In der Regel finden sich keine Zeichen der Mangelernährung. Die gastrointestinalen Beschwerden können die einzige Manifestation sein. Pathophysiologisch spielen die Small-Fiber-Neuropathie mit gestörtem autonomen Nervensystem, Entzündungsprozesse, Dysbiose, Maldigestion und gastrointestinale Ischämien eine Rolle.
HNO
Der sensorineurale Hörverlust tritt häufig auf, betrifft insbesondere hohe Frequenzen und schreitet meist fort. Dabei handelt es sich um einen überwiegend cochleären Hörverlust. Etwa 30 % der Patienten berichten einen akuten, sich über wenige Stunden bis Tage entwickelnden Hörverlust.
Ein Tinnitus ist ein frühes und häufiges Symptom des Morbus Fabry. Weiterhin geben bis zu 60 % der Patienten Schwindel an. Die Ätiologie (vestibulär, zentral oder kardial) ist variabel.
Ophthalmologisch
Ophthalmologische Veränderungen betreffen die Hornhaut, die Linse und die Gefäße von Konjunktiva und Retina. Diese Veränderungen führen meist nicht zu einer Visuseinschränkung.
Die häufigste Manifestation ist die Cornea verticillata, eine wirbelförmige, unterhalb des Hornhautzentrums lokalisierte oberflächliche Trübung der Hornhaut, die bei 40 bis 90 % der Patienten vokommt. Sie kann mittels Spaltlampenuntersuchung diagnostiziert werden. Eine diffuse Trübung des Hornhautstromas wird als "Corneal Haze" bezeichnet.
Ein Morbus Fabry ist mit einer Katarakt assoziiert, wobei die anteriore Form von der posterioren subkapsulären Fabry-Katarakt unterschieden wird. Letztere gilt als erkrankungsspezifiisch, kann aber auch bei anderen lysosomalen Speicherkrankheiten auftreten.
Das Auftreten von geschlängelten Augengefäßen (Tortuositäten) der Retina und Konjunktiva ist eine weitere mögliche Manifestation.
Dermatologisch
Diagnostisch hinweisend auf einen Morbus Fabry sind Angiokeratome. Dabei handelt es sich um meist asymptomatische, kleine, meist gruppiert auftretende, teils hyperkeratotische, rötlich-bräunliche Gefäßerweiterungen. Sie treten bevorzugt in der Leiste, gluteal, periumbilikal, skrotal und an den Oberschenkeln auf. Auch an Händen, Füßen ode Schleimhäuten können sie vorkommen.
Die ersten Angiokeratome lassen sich im Kindesalter nachweisen und nehmen im Laufe des Lebens an Zahl zu. Weiterhin werden asymptomatische Teleangiektasien beobachtet.
Die Hypo- bzw. Anhidrose wird durch eine autonome Dysfunktion sowie durch eine Akkumulation von Gb3 in Schweißdrüsen erklärt. Sehr selten findet sich eine Hyperhidrose.
Weitere Manifestationen
Weitere seltene Symptome sind:
- Lymphödeme, insbesondere der unteren Extremität
- obstruktive Ventilationsstörung mit Bronchialwandverdickungen
- Osteopenie, Osteoporose
- Hüftkopfnekrose
- Anämie
Diagnostik
Bei Männern ist die Bestimmung der alphaGalA-Aktivität im EDTA- oder Heparinblut die Methode der Wahl. Dabei zeigt sich eine pathologisch reduzierte Aktivität der alpha-Galaktosidase A von 0 bis 24 % des unteren Referenzwertes. Der molekulargenetische Nachweis der krankheitsverursachenden Genvariante ist zum Ausschluss falsch-positiver Ergebnisse notwendig. Wenn die AGLA-Aktivität im Normalbereich liegt, ist bei Männern ein Morbus Fabry ausgeschlossen.
Frauen weisen in 20 bis 30 % der Fälle eine normale Ezymaktivität im Blut auf. Daher ist zwingend der Variantennachweis im GLA-Gen zur Diagnosestellung erforderlich. Die Bestimmung der individuellen Genvariante ist weiterhin für die Abschätzung der Pathogenität, der Krankheitsschwere und des Ausmaßes der Organbeteiligung hilfreich.
Werden Organbiopsate durchgeführt (z.B. zur Abklärung einer Kardiomyopathie), so finden sich in Semidünnschnitten oder in der Elektronenmikroskopie zwiebelschalenartige, intrazelluläre Gb3-Akkumulate.
Aktuell (2024) ist der Morbus Fabry noch kein Bestandteil des Neugeborenenscreenings.
Kardio-MRT
Zur Beurteilung der kardialen Manifestationen dient neben der Echokardiographie insbesondere die Kardio-MRT. Dabei zeigen sich folgende Befunde:
- Cine-SSFP: linksventrikuläre Hypertrophie, erhöhte linksventrikuläre Masse, verdickte Papillarmuskeln
- Late Enhancement: fokales mid-myokardiales Late Enhancement (Inflammation und Fibrose), insbesondere basal inferolateral
- T1-Mapping: verminderte T1-Werte (intrazelluläre Akkumulation von Glykosphingolipiden), erhöht in Arealen der Inflammation und Fibrose.
- T2-Mapping: erhöht in Arealen der chronischen Inflammation
- EZV: erhöht in Arealen der chronischen Inflammation
Verlaufskontrollen
Manifestationen wie Nephropathie, kardiale und zerebrovaskuläre Erkrankungen sollten im Rahmen von 12-monatigen Verlaufsuntersuchungen kontrolliert werden. Dabei kann das Lyso-Gb3 im Serum als potenzieller Biomarker zur Beurteilung der Krankheitsprogression bestimmt werden.
Differenzialdiagnosen
Aufgund des variablen klinischen Bildes existieren eine Vielzahl an möglichen Differenzialdiagnosen. Häufige Fehldiagnosen sind:
- andere lysosomale Speicherkrankheiten (z.B. Fukosidose, Sialidose, beta-Mannosidose)
- Wachstumsschmerzen
- Reizdarm-Syndrom
- Morbus Menière
- Morbus Osler
- rheumatische Erkrankungen
- entzündliche ZNS-Erkrankungen (z.B. multiple Sklerose)
Therapie
Patienten mit Morbus Fabry sollten zusätzlich zur ambulanten Behandlung an einem interdisziplinären Fabry-Zentrum vorgestellt werden. Die Erkrankung wird mit einer Enzymersatztherapie behandelt, die einmal alle zwei Wochen durchgeführt wird. Zugelassen sind:
- Agalsidase alfa (Replagal®) i.v. 0,2 mg/kgKG[3]
- Agalsidase beta (Fabrazyme®) i.v. 1,0 mg/kgKG[4]
- Pegunigalsidase alfa (Elfabrio®) i.v. 1 mg/kgKG[5]
Hinweis: Diese Dosierungsangaben können Fehler enthalten. Ausschlaggebend ist die Dosierungsempfehlung in der Herstellerinformation.
Für einige Missense-Mutationen mit noch erhaltener Enzym-Restfunktion ist es außerdem möglich, das defekte Enzym mithilfe des pharmakologischen Chaperons Migalastat (1-Deoxygalactonojirimycin) zu stabilisieren. Jede Kapsel enthält 123 mg Migalastat. Die Einnahme einer Kapsel erfolgt per os jeden zweiten Tag jeweils zur gleichen Uhrzeit mindestens zwei Stunden vor und nach einer Mahlzeit.[6]
Hinweis: Diese Dosierungsangaben können Fehler enthalten. Ausschlaggebend ist die Dosierungsempfehlung in der Herstellerinformation.
Begleitend werden die Organmanifestationen symptomatisch behandelt. Beispielsweise können Schmerzen prophylaktisch mittels Antikonvulsiva wie Carbamazepin behandelt werden. Wenn der betroffene Patient eine Niereninsuffizienz entwickelt hat, können eine Nierendialyse und eventuell auch eine Nierentransplantation notwendig werden.
Asymptomatische Personen mit einer krankheitsverursachenden GLA-Variante sollten alle 12 Monate kontrolliert werden. Die Indikation zur Therapie wird individuell gestellt.
Prognose
Unbehandelt führt der Morbus Fabry zur Einschränkung der Lebensqualität und Lebenserwartung bei beiden Geschlechtern. Hauptursachen für die Morbidität und Mortalität sind renale, kardiale und zerebrovaskuläre Manifestationen (Nieren- und Herzversagen sowie Schlaganfälle). Unbehandelt wird von einer Reduktion der Lebenserwartung um etwa 20 Jahre bei Männern und um 10 Jahre bei Frauen ausgegangen.
Leitlinie
- Üçeyler N. et al DGN S1-Leitlinie Morbus Fabry, Stand 10/2022
Literatur
- aerzteblatt.de – Morbus Fabry – oft gesehen, selten erkannt, abgerufen am 6.6.2023
- orpha.net – Fabry-Syndrom, abgerufen am 6.6.2023
Quellen
- ↑ Spada M et al. High incidence of later-onset fabry disease revealed by newborn screening. Am J Hum Genet. 2006
- ↑ Hwu WL et al. Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c.936+919G>A (IVS4+919G>A). Hum Mutat. 2009
- ↑ Replagal. EMA, abgerufen am 25.02.2024
- ↑ Fabrazyme. EMA, abgerufen am 25.02.2024
- ↑ Elfabrio. EMA, abgerufen am 25.02.2024
- ↑ Galafold. EMA, abgerufen am 25.02.2024
Weblink
- Soloway S, Lister D. Fabry's Disease. N Engl J Med. 2024 - Fallbericht mit Abb.