Hereditäre hämorrhagische Teleangiektasie








Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
Loslegennach dem kanadischen Arzt William Osler (1849-1919), dem französischen Arzt Henri Jules Louis Marie Rendu (1844-1902) und dem englischen Arzt Frederick Parkes Weber (1863-1962)
Synonyme: Morbus Rendu-Osler-Weber, Morbus Rendu-Osler, Morbus Osler, Telangiektasia hereditaria hemorrhagica, Osler-Weber-Rendu-Krankheit, Osler-Weber-Rendu-Syndrom
Abkürzung: HHT
Englisch: Rendu-Osler-Weber disease
Definition
Die hereditäre hämorrhagische Teleangiektasie, kurz HHT, ist eine autosomal-dominant vererbte Vaskulopathie, die zu einer pathologischen Erweiterung der Blutgefäße, sogenannten Teleangiektasien führt.
Sorry, nur für Fachkreise. Wenn du diesen Inhalt sehen willst, musst du eingeloggt sein und auf diesen Link klicken
Zum Inhalt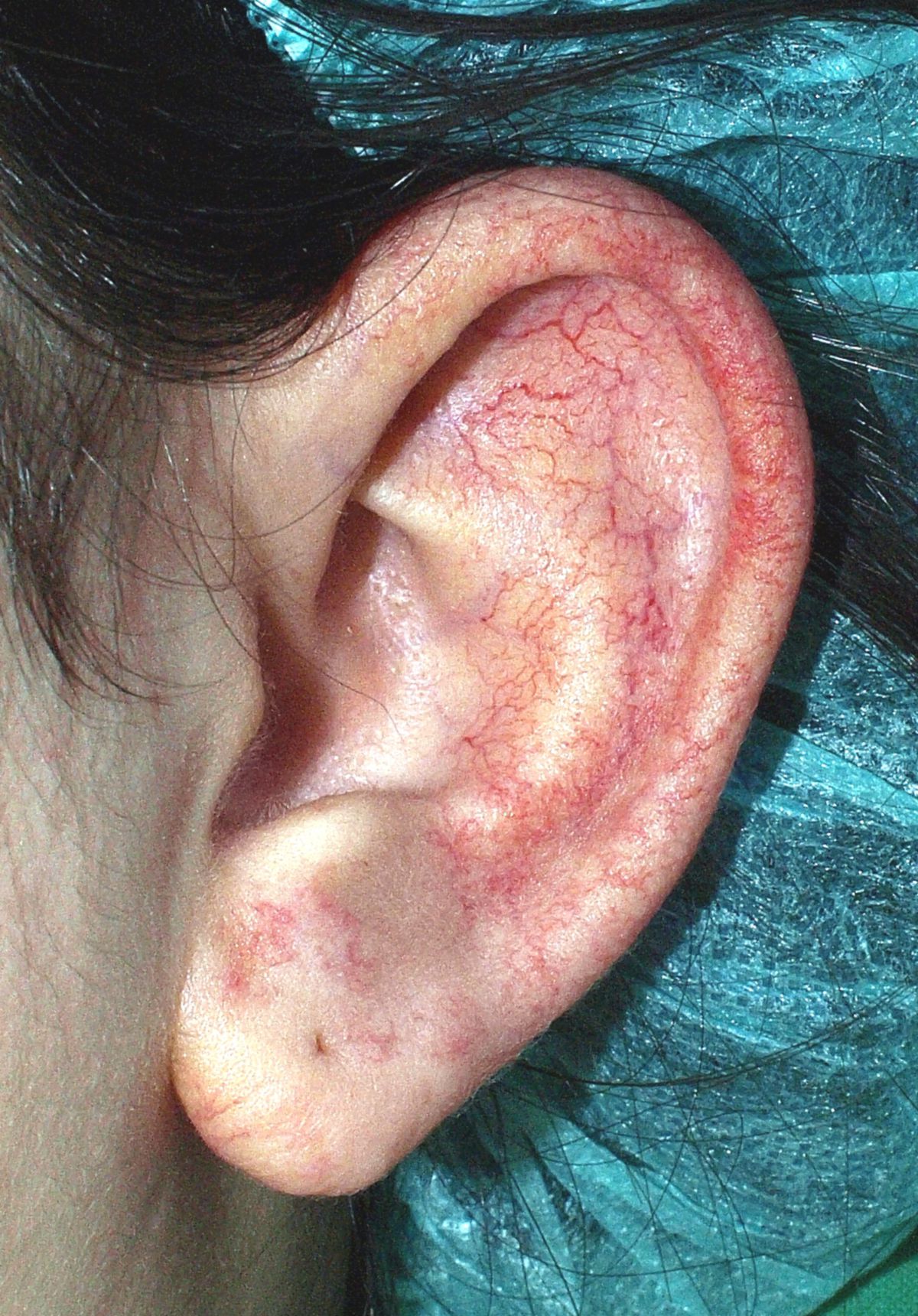
Sorry, nur für Fachkreise. Wenn du diesen Inhalt sehen willst, musst du eingeloggt sein und auf diesen Link klicken
Zum InhaltGenetik
Der Vererbungsgang der HHT ist autosomal-dominant. Bisher sind fünf verschiedene genetische Typen bekannt, von denen drei bestimmten Gendefekten zugeordnet werden können, bei zweien ist lediglich der Genlokus bekannt.
| Typ | Genlokus | Gen |
|---|---|---|
| HHT1 | 9q34.1 | ENG |
| HHT2 | 12q11-q14 | ACVRL1 |
| HHT3 | 5q31 | unbekannt |
| HHT4 | 7p14 | unbekannt |
| HHT5 | 10q11 | GDF2 |
| JPHT | 18q21.1 | SMAD4 |
Bei der HHT1 betrifft der Gendefekt das membranständige Glykoprotein Endoglin, einen Teil des TGF-beta-Rezeptors. Es spielt eine wichtige Rolle bei der Angiogenese. Die HHT2 basiert auf einem Defekt der Activin Receptor-Like Kinase 1, die ebenfalls ein Membranrezeptor für Liganden aus der TGF-beta-Familie ist.
HHT1 geht vermehrt mit vaskulären Malformationen von Lunge und Gehirn einher, während diese bei HHT2 vor allem in der Leber auftreten. Besonders bei HHT2 kann es in seltenen Fällen zu einer pulmonal-arteriellen Hypertonie (PAH) kommen.
Die JPHT ist ein Sonderfall, da die HHT mit einer juvenilen Polyposis kombiniert ist. Das defekte Gen SMAD4 kodiert für einen Transkriptionsfaktor, der auf dem durch TGF-beta aktivierten SMAD-Signalweg liegt.
Pathophysiologie
Bei der HHT entstehen Gefäßerweiterungen mit postkapillärer Dilatation von Venolen und einer Abnahme der umgebenden Kapillaren. Im Verlauf entwickeln sich direkte Kurzschlüsse zwischen Arterien und Venen, welche als Teleangiektasien bzw. als vaskuläre Malformationen in Erscheinung treten.
Epidemiologie
Die Angaben über die Prävalenz schwanken zwischen 1/50.000 bis 1/2.700 Einwohnern.
Klinik
Lunge
Pulmonale arteriovenöse Malformationen (PAVM) kommen bei 10 - 50 % der Betroffenen vor. Oft verlaufen sie lange asymptomatisch oder führen zu Kopfschmerzen. Seltenere Komplikationen sind paradoxe Embolien mit entsprechenden Infarkten (insbesondere Hirninfarkte) und Abszedierungen (Gehirn, Leber, Milz). PAVMs können, insbesondere im Rahmen von Schwangerschaften, zu potenziell fatalen Blutungen mit Hämoptysen oder Hämatothorax führen.
Eine weitere Lungenbeteiligung ist die pulmonale Hypertonie, die sekundär infolge einer Leberbeteiligung oder selten primär als pulmonal-arterielle Hypertonie auftreten kann.
ZNS
Die meisten neurologischen Symptome bei HHT-Patienten entstehen in Folge einer paradoxen Embolie durch PAVMs. Zerebrale vaskuläre Malformationen (CVM) kommen bei 10 bis 23 % der Patienten vor. Dazu zählen arteriovenöse Malformationen, AV-Fisteln und Teleangiektasien. Das Risiko für symptomatische Blutungen ist vermutlich geringer als bei sporadischen CVM.
Spinale vaskuläre Malformationen sind deutlich seltener.
Leber
Hepatische vaskuläre Malformationen (HVM) kommen bei 32 bis 78 % der Fälle vor, werden jedoch nur in ca. 8 % symptomatisch. Mögliche Manifestationen sind:
- hyperdynames Herzversagen mit Leistungsabnahme und Dyspnoe (durch eine hämodynamisch relevante Shuntmenge)
- portale Hypertension, teils mit gastrointestinalen Blutungen oder Aszites
- biliäre Verlaufsform mit Ikterus und Bauchschmerzen.
- sekundäre pulmonale Hypertonie
- fokal noduläre Hyperplasie (FNH)
Gastrointestinaltrakt
Teleangiektasien der gastrointestinalen Schleimhäute oder Angiodysplasien treten bei bis zu 91 % der Betroffenen auf, bluten aber nur in ca. 30 % der Fälle und selten vor dem 40. Lebensjahr. Schwarzer verfärbter Stuhl tritt häufiger durch Schlucken von Blut aus der Nase auf.
Epistaxis
Epistaxis ist das häufigste Symptom und lässt sich bei ca. 95 % der Betroffenen nachweisen, erstmals meist im Pubertätsalter. Beginn und Verlauf sind jedoch sehr variabel.
Diagnostik
Die Verdachtsdiagnose kann in der Regel bereits aufgrund der Anamnese und der typischen Klinik gestellt werden. Ergänzend ist eine molekulargenetische Diagnostik zum Nachweis eines Gendefekts sinnvoll.
Bei allen Patienten mit mutmaßlicher oder nachgewieser HHT sollte ein Screening auf eine PAVM erfolgen. Methode der Wahl ist die kontrastmittelgestützte Echokardiographie, als Alternative steht die CT zur Verfügung. Eine Wiederholung der Untersuchung wird nach circa fünf Jahren empfohlen.
Zum Nachweis von zerebralen Gefäßfehlbildungen ist die MRT die Methode der Wahl. Ob und wann ein Screening auf CVM mittels MRT notwendig ist, ist derzeit (2025) Gegenstand von Diskussionen.
Die Diagnose hepatischer Manifestationen erfolgt üblicherweise mit Dopplersonografie, gegebenenfalls mit kontrastmittelgestützter CT oder MRT. Biopsien sollten vermieden werden.
Ab dem 35. Lebensjahr sollte mindestens einmal jährliche eine Bestimmungen der Hämoglobinkonzentration erfolgen. Bei einer Diskrepanz zur Epistaxis ist eine Endoskopie des Magen-Darmtraktes sinnvoll.
Diagnosekriterien
Die Diagnosekriterien sind in den sogenannten Curaçao-Kriterien festgelegt:[1]
- Heredität: Dieses Kriterium ist bei wenigstens einer verwandten Person ersten Grades mit gesicherter Diagnose erfüllt.
- Hämorrhagie/Epistaxis: Bedingung für dieses Kriterium ist rezidivierendes, spontanes Nasenbluten.
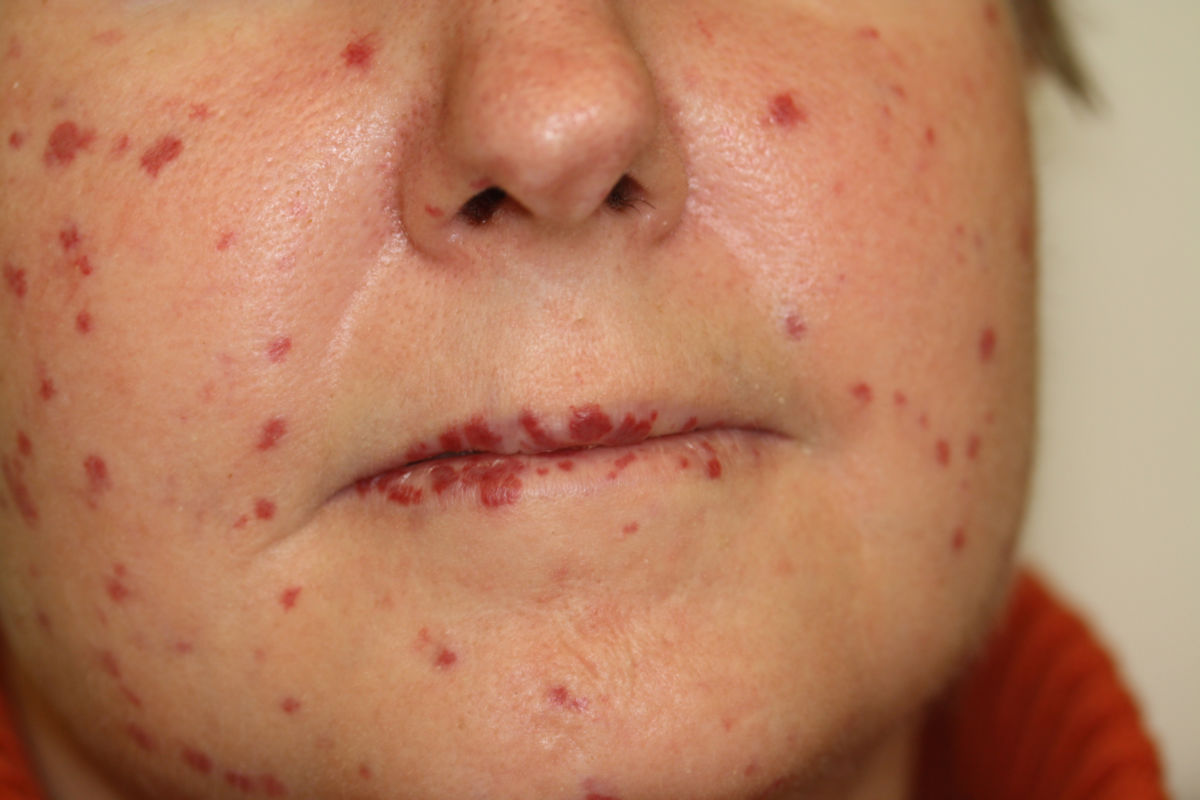
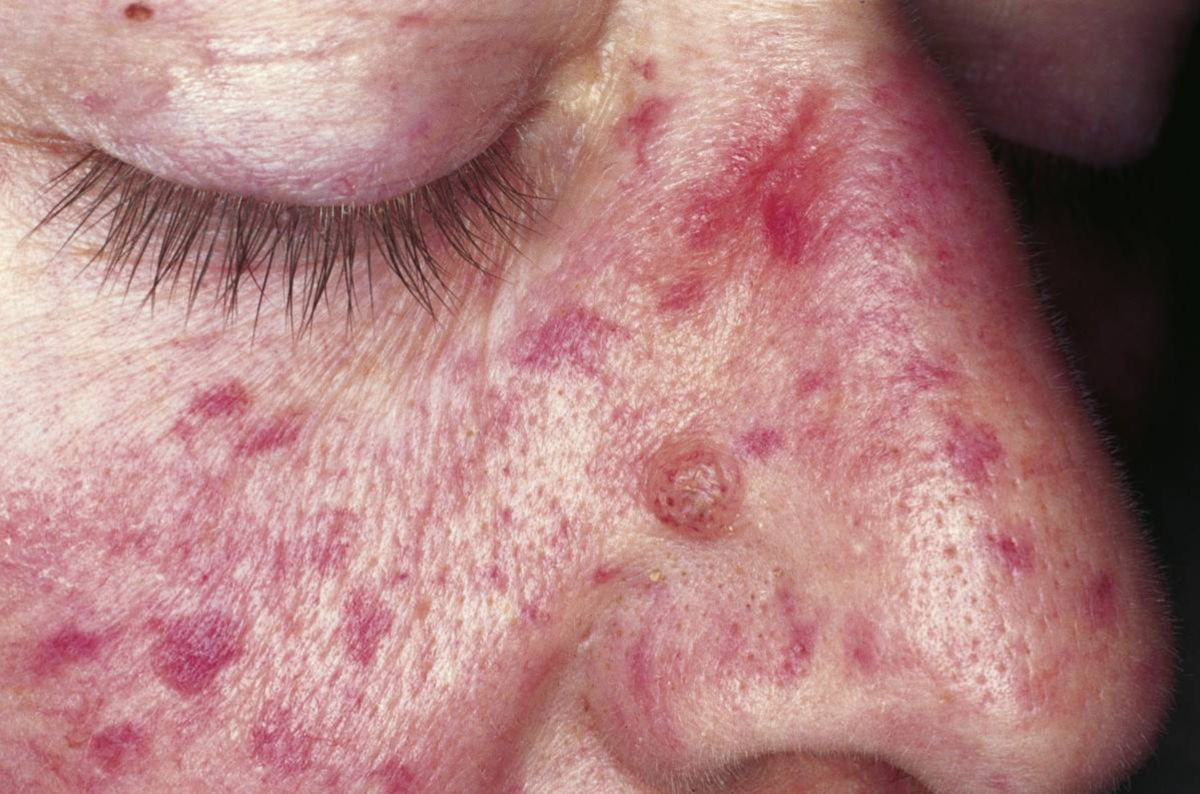
- Teleangiektasien: in typischen Prädilektionsstellen (Mundschleimhaut, Lippe, Finger, Nasenschleimhaut)
- Organbeteiligung; hierzu zählen:
- zerebrale vaskuläre Malformationen (CVM)
- hepatische vaskuläre Malformationen (HVM)
- pulmonale arteriovenöse Malformationen (PAVM)
- gastrointestinale Beteiligung
Wenn 2 dieser 4 Kriterien erfüllt sind, ist das Vorliegen einer HHT möglich, ab 3 gilt sie nach dieser Klassifikation als diagnostiziert.
Therapie
Die Behandlung unterscheidet sich je nach Manifestation:
Epistaxis
Die Therapie erfolgt basierend auf einem Stufenkonzept: Intial steht zunächst die Befeuchtung der Nasenschleimhaut im Vordergrund. Möglich sind Inhalationen, Öle, Salben, Gele, Spülungen und Raumluftbefeuchter. Propranolol oder Tacrolimus können off label Salben und Gelen beigemischt werden. Die Betroffenen sollten geschult werden, wie sie ihre Nase von außen komprimieren können. Hierbei kann auch ein Nasenclip hilfreich sein. Auch das selbstständige Anlegen einer Tamponade ist sinnvoll.
Als nächste Stufe erfolgt die orale Gabe von Tranexamsäure (bis dreimal 1 g) oder die orale Gabe von N-Acetylcystein (3-mal täglich 600 mg, off label). Möglich ist auch eine endonasale Koagulation mittels Nd:YAG-Laser oder die endonasale Sklerosierung durch lokale Injektion. Letztere Therapieoption ist effizient, jedoch aufgrund des seltenen Risikos einer Erblindung umstritten.
Als dritte Stufe kommt eine antiangiogenetische Therapie (off label) in Frage:
Operative Behandlungen der dritten Stufe sind die Septodermoplastik nach Saunders und der Nasenverschluss nach Young. Bei der Septodermoplastik wird die betroffene Nasenschleimhaut entfernt und anschließend durch ein Schleim-/Hauttransplantat ersetzt. Zu den Nebenwirkungen gehören ausgeprägte Verkrustungen der Nase, Ozaena („Stinknase“), Nasenatmungsbehinderung, Riechverlust und das Wiederauftreten von Blutungen in schwerer zu behandelnden Bereichen. Im Gegensatz zur eher seltener durchgeführten Septodermoplastik ist der komplette Nasenverschluss meist umkehrbar. Nachteilig sind die notwendige Mundatmung und der Riechverlust. Es ist jedoch bisher die einzige Methode, welche dauerhaft zu einem kompletten Sistieren der Epistaxis führen kann. Sie ist auch effektiv, wenn eine Antikoagulation oder Thrombozytenaggregationshemmung notwendig ist.
Lunge
Behandlungsmethode der Wahl bei PAVM ist die perkutane Katheterembolisation. Nur sehr selten ist eine chirurgische Resektion sinnvoll.
Solange eine PAVM nicht sicher ausgeschlossen wurde, empfehlen viele Autoren die Gabe einer prophylaktischen Antibiose bei allen Eingriffen mit potentieller Bakteriämie entsprechend den aktuellen Endokarditis-Richtlinien.
ZNS
Meist ist eine konservative Therapie ausreichend, jedoch sollte die Wahl der Behandlung interdisziplinär in einem Zentrum mit entsprechender Expertise diskutiert werden.
Leber
Die Behandlung der symptomatischen Leberbeteiligung ist komplex. Insbesondere beim hyperdynamen Herzversagen kommen Betablocker und Diuretika, bei Therapieversagen Bevacizumab (off label) in Frage. Bei rascher Progredienz kann eine zeitnahe Lebertransplantation erforderlich sein. Selten werden Leberembolisationen durchgeführt.
Gastrointestinaltrakt
Argon-Plasma-Koagulationen können bei gastrointestinalen Teleangiektasen anfangs versucht werden. Bleibt der Erfolg aus, kommen Antifibrinolytika wie Tranexamsäure (off label) und Hormone (Östrogen-Gestagentherapie) in Frage. Bei Therapieversagen kann Bevacizumab (off label) erwogen werden.
Antikoagulation
Blutungen bei HHT stellen keine absolute Kontraindikation für eine Antikoagulation oder Thrombozytenaggregationshemmung dar. Eine doppelte Thrombozytenaggregationshemmung oder die Kombination mit Antikoagulation sollte möglichst vermieden werden.
Schwangerschaft
Schwangere mit HHT mit unbehandelten CVM oder PAVM oder nicht kürzlich mittels Screening ausgeschlossener PAVM werden als Risikoschwangerschaft eingestuft.
Selbsthilfegruppen
Für Betroffene ist der Kontakt zu Selbsthilfegruppen oft hilfreich:
Quelle
- ↑ Shovlin CL et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet. 2000