Sarkoidose









Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenSynonyme: Morbus Boeck (sprich: buhk), Morbus Besnier-Boeck-Schaumann, Morbus Schaumann-Besnier
Englisch: sarcoidosis
Definition
Die Sarkoidose ist eine granulomatöse Entzündung. Sie kann prinzipiell jedes Organ befallen, fällt klinisch jedoch am ehesten durch den Befall der Lungen auf.
Epidemiologie
Die Sarkoidose hat eine Inzidenz von 10 bis 40 Neuerkrankungen pro 100.000 Einwohner und manifestiert sich bevorzugt zwischen dem 2. und 4. Lebensjahrzehnt. Spitzen sind jedoch vor allem zwischen dem 20. und 30. sowie dem 60. und 70. Lebensjahr und insbesondere bei Frauen zu finden. Das Mann-zu-Frau-Verhältnis liegt etwa bei 2:1.
Vergleichsweise besonders häufig tritt die Sarkoidose in Schweden, Dänemark und bei Afroamerikanern auf. In Mittelmeerländern und Afrika ist die Sarkoidose eine Rarität. Erstaunlicherweise sind Raucher seltener von einer Sarkoidose betroffen als Nichtraucher.
Pathologie
In den befallenen Organen findet man typische Granulome, die im Gegensatz zu den besser bekannten Granulomen der Tuberkulose keine zentrale Nekrose und somit auch keine Verkäsung aufweisen.
Pathognomonisch für die Granulome der Sarkoidose sind zwei lichtmikroskopische Zeichen:
Auf makroskopischer Ebene äußert sich die Sarkoidose in einem variablen Muster des Organbefalls. Typische Manifestationen sind:
- Lymphknotensarkoidose: Die Lymphknoten sind fast immer betroffen, insbesondere die am Hilus der Lungen gelegenen thorakalen Lymphknoten. Klinisches Korrelat ist die bihiläre (beide Hili betreffende) Lymphadenopathie.
- Lungensarkoidosen: In über 70 % der Fälle sind die Lungen (Interstitium) betroffen. Die Sarkoidose kann bei ungünstigem Verlauf zur Lungenfibrose führen.
- Augensarkoidose: In ca. einem Fünftel der Fälle sind die Augen mit Uveitis, Iridozyklitis, Kalkablagerungen in Binde- und Hornhaut sowie einer granulomatösen Entzündung der Tränendrüse betroffen.
- Hautsarkoidose: Das Erythema nodosum ist eine vorzugsweise über der Vorderseite der Tibia auftretende, häufig schmerzhafte Veränderung der Haut mit rötlichen Papeln. Eine weitere Hautmanifestation äußert sich als sogenannter Lupus pernio.
- Neurosarkoidose: Im ZNS kann eine granulomatöse Enzephalitis auftreten, die durch Druck auf Hirnstrukturen beispielsweise eine Fazialisparese verursachen kann. Auch ein Diabetes insipidus kann durch den ZNS-Befall ausgelöst werden.
- Knochensarkoidose: Am Knochen kann die Sarkoidose zystische Veränderungen der Fingerknochen verursachen, die typischerweise im Rahmen des Jüngling-Syndroms auftreten.
- Milzsarkoidose und Lebersarkoidose: Die Milz und die Leber zeigen bei Befall charakteristische Granulome.
- Herzsarkoidose: Bei Befall des Herzens können im ungünstigen Fall Teile des Erregungsleitungssystems von Granulomen befallen werden. Potentiell tödliche Herzrhythmusstörungen sind die Folge.
- Nierensarkoidose: Im Rahmen einer Hyperkalzämie, die bei Sarkoidose in ca.15 % der Fälle auftritt, findet sich eine Hyperkalziurie in den Nieren, die Nierensteinbildung wird dadurch begünstigt.
- Speicheldrüse: Eine Sarkoidose der Parotis tritt im Rahmen des Heerfordt-Syndroms auf.
Leber und Skelettmuskulatur können ebenfalls granulomatöse Entzündungsherde aufweisen.
Histopathologie
Ätiologie
Die genaue Ätiologie der Sarkoidose ist unbekannt. Derzeit (Stand 2026) werden genetische, immunologische und infektiöse Ursachen diskutiert.
Die Granulome werden als Reaktion auf ein bisher unbekanntes Antigen gedeutet. Dafür spricht, dass an betroffenen Stellen die Konzentration von Zytokinen wie Interleukin-2 und Interferon-gamma gesteigert sind. Beide Zytokine sind charakteristisch für die Aktivität von CD4-positiven T-Helferzellen vom Typ 1 und locken CD8-positive T-Lymphozyten (zytotoxische T-Lymphozyten) und Makrophagen zum Ort des Geschehens. Der CD4/CD8-Quotient ist in betroffenen Organen erhöht.
Durch die saisonale Häufung der Krankheitsfälle wird ein infektiöser Auslöser, unter anderem Bakterien, diskutiert. Bisher liegen jedoch keine beweisenden Erkenntnisse zu diesem Punkt vor. Im Gegensatz dazu konnte jedoch eine erhöhte Erkrankungswahrscheinlichkeit bei Individuen mit bestimmten HLA-Genotypen (HLA-DQB1) festgestellt werden. Auch andere Forschungsergebnisse untermauern die genetische Ätiologie. So kann die Mutation eines einzigen Basenpaares im Gen BTNL2 auf dem Chromosom 6 die Erkrankungswahrscheinlichkeit um ca. 60 % zu erhöhen.
Symptomatik
Die Sarkoidose kann akut oder chronisch auftreten. Beide Formen unterscheiden sich in ihren Symptomen. Für den akuten Verlauf spricht das Vorliegen von
- Fieber
- Arthralgien
- Erythema nodosum
- trockener Husten
- Dyspnoe bei Belastung
- Vergrößerung mediastinaler Lymphknoten
Mögliche Symptomkomplexe der akuten Sarkoidose sind:
Der chronische Verlauf äußert sich über Monate einschleichend in
- langsam zunehmender Dyspnoe, insbesondere bei Belastung
- (länger bestehendem) trockenem Husten
- Arthralgien, deren Intensität unvorhersehbar zu- oder abnimmt
- Synechien im Auge: Synechie der Regenbogenhaut mit der davorliegenden Hornhaut oder der dahinterliegenden Augenlinse
Eine seltener Symptomkomplex der chronischen Sarkoidose ist das Jüngling-Syndrom.
Diagnostik
Bei Verdacht auf Sarkoidose sollte eine konventionelle Röntgenaufnahme des Thorax aufgenommen und eine Lungenfunktionsuntersuchung durchgeführt werden. Bei der Lungenfunktionsuntersuchung ist eine restriktive Ventilationsstörung festzustellen.
Bildgebung
Röntgen-Thorax
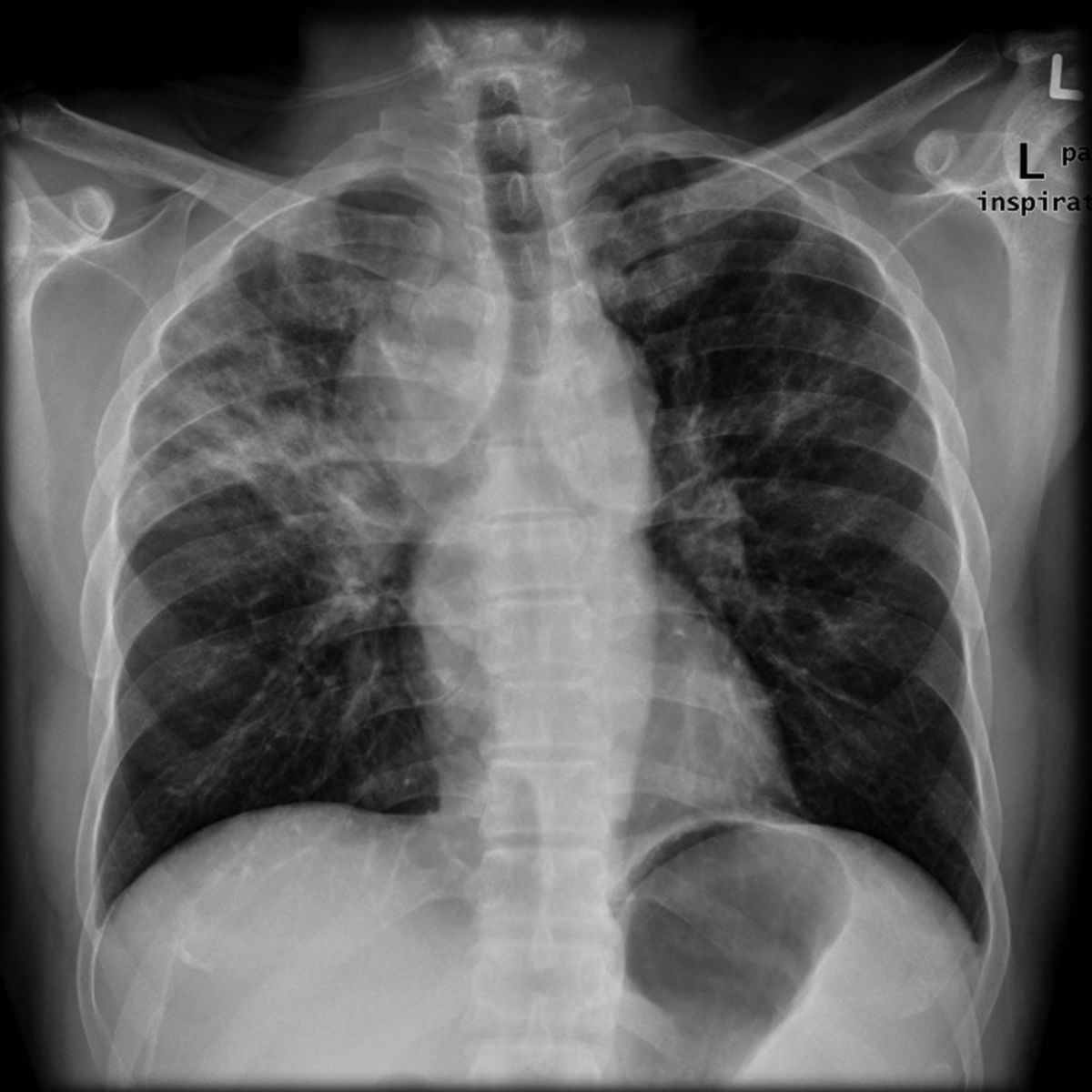
Das Röntgenbild zeigt typische Veränderungen, die auch zur Einteilung der Sarkoidose nach Scadding genutzt werden. Dabei handelt es sich um unterschiedliche Typen, nicht um Stadien, die ineinander übergehen:
- Typ 0: unauffälliger Thorax bei extrapulmonalem Befall
- Typ 1: bihiläre Lymphadenopathie ohne sichtbare Lungenbeteiligung, Hilusvergrößerung in der Regel beidseits
- Typ 2: bihiläre Lymphadenopathie mit Lungenbeteiligung, die Lunge zeigt eine retikulonoduläre Zeichnung
- Typ 3: Lungenbefall ohne Fibrose und ohne sichtbare Lymphadenopathie
- Typ 4: Lungenfibrose
Eine ältere Klassifikation nach Wurm unterteilt die Sarkoidose in 3 Stadien.
HR-CT
Eine differenziertere Betrachtung der Parenchymveränderungen ist mit Hilfe der hochauflösenden Computertomographie möglich. Die Einteilung nach Scadding sollte hierbei nicht angewendet werden. Typische Befunde sind:
- Lymphadenopathie: Sie tritt mediastinal und bihilär symmetrisch auf. Initial sind die Lymphknoten in 5 % der Fälle verkalkt, im weiteren Verlauf in bis zu 50 % der Fälle (Puderzucker-artig ("icing sugar"), Eierschalenverkalkung, grobschollig).
- Pulmonale Noduli: Solide, < 1 mm bis 1 cm, teilsweise auch größer, perilymphatische Verteilung, betont zentral bzw. perihilär sowie in Ober- und Mittelfeld.
- Sie sind teilweise so fein, dass sie wie Milchglastrübungen imponieren oder subpleural zu Pseudoplaques akkumulieren.
- Galaxy-Zeichen: Dichter Kern aus akkumulierten Noduli mit umgebenden feinen Noduli. Die Konstellation erinnert morphologisch an eine Galaxie. Zentrale Kavitäten und umgebende Milchglastrübungen (durch mikroskopisch kleine Noduli) können auftreten.
- Lungenfibrose: Sie tritt in 20-25 % der Fälle auf und muss nicht mit Noduli kombiniert sein. Typischerweise kommt es zu einer konglomeratartigen Fibrose mit dominierenden Traktionsbronchiektasen und Architekturstörungen. Sie treten betont perihilär im Mittel- und Oberfeld auf. Teilweise besteht auch ein komplett unspezifisches Fibrosemuster.
Bronchoskopie
Bei röntgenologischem Verdacht auf Sarkoidose dient die Bronchoskopie mit bronchoalveolärer Lavage zur Bestätigung. In der Lavageflüssigkeit zeigt sich ein erhöhter CD4/CD8-Quotient. Bei einem Quotienten > 5 ist eine Sarkoidose wahrscheinlich. Der CD4/CD8 Quotient dient insbesondere zur Differentialdiagnose einer Lungenfibrose, bei welcher der Quotient normal wäre.
Alternativ kann auch eine Mediastinoskopie durchgeführt werden, um direkt die Hiluslymphknoten zu untersuchen. Diese ist jedoch mit einem höheren Risiko für den Patienten verbunden.
Neuere Studien zeigen, dass die transbronchiale Nadelaspiration (TBNA) der Lymphknoten im Rahmen des endobronchialen Ultraschalls (EBUS) eine höhere Treffsicherheit zum Nachweis von Granulomen beinhaltet als die konventionelle Bronchoskopie. Risiken eines Pneumothoraxes oder Hämorrhagien wie bei der transbronchialen Lungenbiopsie können so vermieden werden.
Labordiagnostik
Die Erythrozytensedimentationsrate (ESR) sowie die BSG sind erhöht, insbesondere bei der akuten Form. Durch die vermehrte Aktivität der T-Helferzellen mit Stimulation von B-Lymphozyten sind in 50 % der Fälle eine Hypergammaglobulinämie vorhanden.
Ein wichtiger diagnostischer Parameter ist die erhöhte Konzentration des löslichen Interleukin-2-Rezeptors α (sIL-2Rα) im Serum, die den Aktivierungsgrad der T-Lymphozyten widerspiegelt. Dieser Parameter ist leicht bestimmbar und weist eine hohe Stabilität und Sensitivität auf. Bei 80 % der Erkrankten lassen sich erhöhte Werte im Serum nachweisen, die jedoch auch im Rahmen chronischer Infektionen, Hämoblastosen sowie bei einigen Tumoren auftreten können.
Zur Verlaufskontrolle der Sarkoidose kann das Angiotensin Converting Enzyme (ACE) im Serum bestimmt werden. Es ist nicht charakteristisch für die Sarkoidose, aber bei vorliegen einer Sarkoidose oft erhöht. Im Rahmen der Sarkoidose wird die Bildung von ACE durch T-Lymphozyten kontrolliert. Die Entzündungsaktivität der Makrophagen lässt sich mit Hilfe des Neopterin-Spiegels erfassen. Im Rahmen der Makrophagenaktivierung findet auch vermehrt die Lysozymfreisetzung statt, was zu entsprechend hohen Serumwerten führt.
Ein weiterer Biomarker ist die Adenosin-Desaminase, welche die zelluläre Immunantwort repräsentiert und bei Entzündungen in erhöhten Mengen produziert wird.
Unspezifisch kann der Serumspiegel an 1,25-Dihydroxycholecalciferol im Rahmen der Sarkoidose erhöht sein.
Differentialdiagnosen
Differentialdiagnostisch kommen (in verschiedenen Stadien) in Frage:
Gerade bei einer Zufallsdiagnose durch einen Röntgenthorax mit der Diagnose einer bihilären Lymphadenopathie (ohne sonstige Beschwerden) ist die Sarkoidose wahrscheinlicher als eine andere Diagnose. Aufgrund ihrer Seltenheit wird die Erkrankung häufig erst nach einem langen diagnostischen Prozess entdeckt.
Prognose
Die Sarkoidose hat in der Regel eine günstige Prognose. Bei der akuten Form kommt es in 90 % der Fälle spontan zur Remission. Im Stadium I nach Scadding findet in 70 % der Fälle einer Spontanheilung statt. Etwa 30 % der Erkrankten leiden nach Abheilung an Einschränkungen der Lungenfunktion bzw. bleibenden Schäden an anderen betroffenen Organen. 10 % der Betroffenen entwickeln eine fortschreitende Lungenfibrose. In wenigen Fällen treten potentiell tödliche Komplikationen wie beispielsweise Herzrhythmusstörungen, terminale Lungenfibrose und Cor pulmonale auf.
Nach Ausbruch ist aber auch ein lebenslanger Verlauf möglich, der unerkannt und asymptomatisch, aber auch symptomatisch sein kann. In letzterem Fall stehen Arthralgien und eine über die Jahre ansteigende Dyspnoe im Vordergrund.
Beteiligungen anderer Organe (Haut, Herz, Gehirn, Leber, usw.) sind jederzeit möglich, weshalb der Patient regelmäßig überwacht werden sollte. Die Hautsarkoidose kann dabei sehr unterschiedliche Erscheinungsformen annehmen.
Therapie
Eine echte Kausaltherapie der Sarkoidose ist zur Zeit (2026) nicht verfügbar. Primär wird die Erkrankung daher zurückhaltend therapiert (Watchful Waiting). Auch nach fünf Jahren ohne Behandlung ist die Mortalität in den Stadien I-III nicht erhöht.[1]
Der Patient sollte in Abständen von 3 Monaten eine Lungenfunktionsuntersuchung und in Abständen von 6 Monaten eine Röntgenuntersuchung des Thorax erhalten. So kann die Sarkoidose unter kontrollierten Bedingungen ggf. von selbst abheilen – bei einer akuten Sarkoidose beträgt die Spontanremissionrate mehr als 80 %. Die symptomatische Behandlung umfasst Analgetika (z.B. Paracetamol, Ibuprofen oder Diclofenac), bei Lungenbeteiligung u.U. auch Antitussiva (z.B. Hydrocodon).
Eine Indikation zu einer immunsuppressiven Therapie besteht bei:
- Vorliegen von Stadium 2 der Erkrankung (s.o.) mit zunehmender Verschlechterung der Lungenfunktion und/oder sehr hohem CD4/CD8-Quotienten.
- Befall extrapulmonaler Organe
- Hyperkalzämie
- klinisch schweren Verläufen
Initial werden Glukokortikoide als Monotherapie über 2 bis 6 Wochen gegeben, anschließend wird die Dosis schrittweise reduziert. Sind symptomatische, behandlungsbedürftige Verläufe unter Steroidtherapie refraktär, wird eine Immunsuppression mit Methotrexat empfohlen. Bei Intoleranz oder Ineffizienz ist ein Wechsel auf Azathioprin oder Hydroxychloroquin, auch in Kombination, möglich. Reicht dies nicht aus, können Infliximab in einer Dosis von 3–5 mg/kgKG alle 6 Wochen i.v. oder Adalimumab verabreicht werden.
Hinweis: Diese Dosierungsangaben können Fehler enthalten. Ausschlaggebend ist die Dosierungsempfehlung in der Herstellerinformation.
Bei Hautsarkoidose konnte in Einzelfällen eine Besserung mit Minocyclin erreicht werden.[2]
Quiz
Bildquelle
- Bildquelle für Flexikon-Quiz: © Vlad Alexandru Popa / pexels
Literatur
- Laborlexikon.de; abgerufen am 21.04.2021
Quellen
- ↑ Hermann, Josef: Sarkoidose. Wie, wann und womit behandeln. Skriptum 2018; 11: 7-9.
- ↑ Sasaki S et al: Management of skin sarcoidosis with minocycline monotherapy. Respirology Case Reports Volume 7, Issue 4 May 2019 Volume 7, Issue 4 May 2019 https://doi.org/10.1002/rcr2.413