Amyloidose







Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
Loslegenvon altgriechisch: ἄμυλον ("ámylon") - Stärke
Englisch: amyloidosis
Definition
Mit dem Begriff Amyloidose fasst man Erkrankungen zusammen, die auf einer Proteinfehlfaltung beruhen. Sie sind durch eine typischerweise extrazelluläre Ablagerung von unlöslichen, polymeren Proteinfibrillen gekennzeichnet, die man Amyloid nennt.
Begriffsherkunft
Der Begriff "Amyloid" wurde im 19. Jahrhundert durch den Pathologen Rudolf Virchow geprägt, der davon ausging, dass die Ablagerungen stärkeartig seien, da sie sich mit Jod-Schwefelsäure blau-violett färbten.
Einteilung
Amyloidosen werden durch die biochemischen Eigenschaften des Proteins, aus dem die Fibrillenablagerungen bestehen, definiert. Außerdem unterscheidet man zwischen systemischen und lokalisierten, erworbenen und hereditären Amyloidosen. Letztere werden auch als AF-Amyloidosen zusammengefasst.
Systemische Amyloidosen
Bei den systemischen Amyloidosen unterscheidet man u.a.:
- AL-Amyloidosen: Immunglobulin-Leichtkette
- AH-Amyloidose: Immunglobulin-Schwerkette
- AA-Amyloidosen: Serumamyloid A
- Aβ2M-Amyloidose: β2-Mikroglobulin
- ATTR-Amyloidose: Transthyretin
- AApoAI-Amyloidose: Apolipoprotein AI
- AApoAII-Amyloidose: Apolipoprotein AII
- AGel-Amyloidose: Gelsolin
- AFib-Amyloidose: Fibrinogen Aα
- ALys-Amyloidose: Lysozym
- ALECT2-Amyloidose: Leukozyten-chemotaktischer Faktor 2
Lokalisierte Amyloidosen
Bei den lokalisierten Amyloidosen differenziert man u.a. zwischen folgenden Formen:
- Aβ-Amyloidose: Amyloid-β-Protein
- ACys-Amyloidose: Cystatin C
- APrP-Amyloidose: Prionprotein
- AIAPP-Amyloidose: Inselzellen-Amyloidpolypeptid (Amylin)
- ACal-Amyloidose: Calcitonin
- AANF-Amyloidose: Atrialer natriuretischer Faktor
- APro-Amyloidose: Prolaktin
- ASgl-Amyloidose: Semenogelin I
- AK-Amyloidose: Keratin
Ätiologie
Die Ursachen der verschiedenen Amyloidoseformen unterscheiden sich erheblich. Der AL-Amyloidose liegt beispielsweise eine monoklonale B-Zell- oder Plasmazellerkrankung zugrunde, die mit einem Myelom oder Lymphom assoziiert sein kann. Die ATTR-Amyloidose wird meist durch Mutation von Transthyretin verursacht. Die AA-Amyloidose tritt sekundär nach chronischer Entzündung oder Infektion sowie bei einem hereditären Fiebersyndrom (z.B. familiärem Mittelmeerfieber) auf.
Pathophysiologie
Die genauen Mechanismen der Fibrillenbildung und der Gewebetoxizität sind noch (2024) unklar. Vermutlich bilden die fehlgefalteten Proteine zunächst oligomere Aggregate, von denen die höchste Toxizität ausgeht. Sie führen zur Bildung von reaktiven Sauerstoffverbindungen. Die Oligomere werden weiterhin zu Polymeren höherer Ordnung und Fibrillen umgewandelt. Letztere lagern sich im Gewebe ab und stören dann die normale Organfunktion.
Klinik
Die klinischen Manifestationen sind vielfältig. Insbesondere bei ungeklärter Nephropathie, Kardiomyopathie (v.a. bei diastolischer Dysfunktion), Neuropathie, Enteropathie, Arthropathie und/oder Makroglossie sollte an eine Amyloidose gedacht werden.
Amyloidosen gehen weiterhin mit relativ unspezifischen Veränderungen in Routinelaboruntersuchungen einher. Das Blutbild ist meist normal, die ESR häufig erhöht. Patienten mit Nierenbeteiligung weisen gewöhnlich eine Proteinurie im nephrotischen Bereich auf, die zu Hypalbuminämie mit Ödemen bzw. Anasarka führen kann.
Die Amyloidkardiomyopathie ist durch eine konzentrische Ventrikelhypertrophie und eine diastolische Funktionsstörung mit erhöhten Serumspiegeln von BNP bzw. NT-proBNP sowie Troponin gekennzeichnet.
Patienten mit Leberbeteiligung entwickeln i.d.R. eine Cholestase mit Anstieg der alkalischen Phosphatase aber nur geringer Transaminasenerhöhung. Bei der AL-Amyloidose kann es bei Infiltration endokriner Organe zu Hypothyreose, Nebennierenrindeninsuffizienz oder Hypopituitarismus kommen.
| Form | Klinisches Syndrom | Manifestationsort |
|---|---|---|
| AL | Primär oder Myelom-assoziiert | Jede möglich |
| AH | Primär oder selten Myelom-assoziiert | Jede möglich |
| AA | sekundär nach chronischer Entzündung oder Infektion, bei hereditären Fiebersyndromen | v.a. Niere |
| Aβ2M | chronische Niereninsuffizienz mit Hämodialyse | Synovialis, Knochen |
| ATTR | Familiär oder altersbedingt | Herz, PNS, ANS |
| AApoAI | Familiär | Leber, Niere |
| AApoAII | Familiär | Niere |
| AGel | Familiär | Kornea, Hirnnerven, Haut, Nieren |
| AFib | Familiär | Nieren |
| ALys | Familiär | Nieren, Leber |
| ALECT2 | Unklar | Nieren |
| Aβ | Alzheimer-Krankheit, Down-Syndrom | ZNS |
| ACys | Zerebrale Amyloidangiopathie | ZNS, Gefäße |
| APrP | Spongiforme Enzephalopathie | ZNS |
| AIAPP | Assoziiert mit Diabetes mellitus | Pankreas |
| ACal | Medulläres Schilddrüsenkarzinom | Schilddrüse |
| AANF | Vorhofflimmern | Herzvorhöfe |
| APro | Endokrine Störung | Hypophyse |
| ASgl | Altersbedingt | Samenblasen |
Diagnostik
Die Diagnose einer Amyloidose beruht auf dem histopathologischen Nachweis der Amyloidablagerung und der immunhistochemischen, biochemischen oder genetischen Analyse des Amyloidtyps.
Histopathologie
Häufig wird Fett der Bauchwand per Nadelaspiration gewonnen und anschließend auf einem Objektträger gestrichen und gefärbt. Bei Patienten mit systemischer Amyloidose lässt sich so in über 80 % d.F. Amyloid nachweisen. Bei negativem Ergebnis können auch Blutgefäße der Gingiva oder der rektalen Schleimhaut sowie Niere, Herz, Leber oder Magen-Darm-Trakt biopsiert werden. Wird die Milz biopsiert, kann sich eine Schinkenmilz oder eine Sagomilz zeigen.
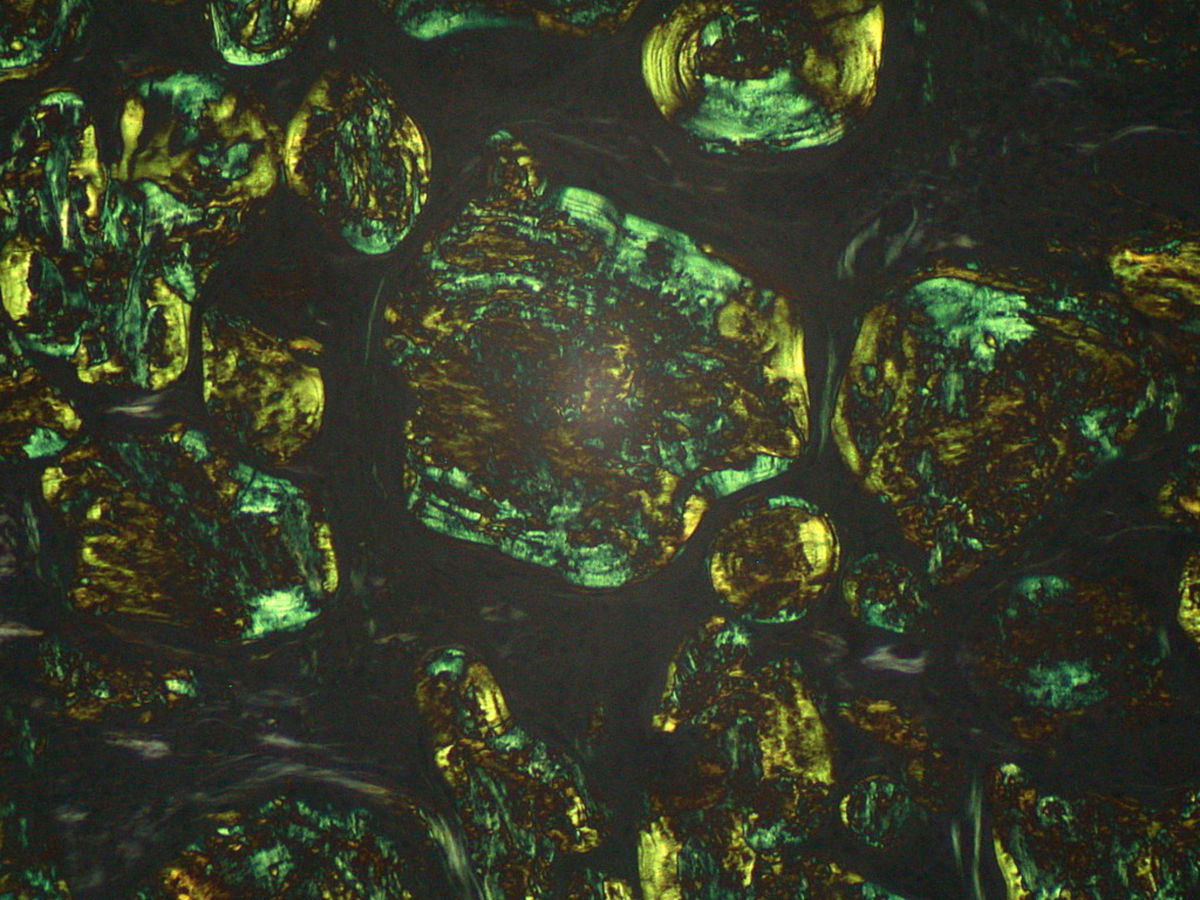
Die β-Faltblatt-Struktur der Amyloidablagerungen führt bei Kongorot-Färbung zu einer grünen Doppelbrechung in der Polarisationslichtmikroskopie. Fibrillen mit 10 nm Durchmesser können auch mit Paraformaldehyd fixiert und direkt mittels Elektronenmikroskopie nachgewiesen werden.
Typisierung
Der Typ des Vorläuferproteins wird anschließend z.B. mittels Immunhistochemie, Immunelektronenmikroskopie oder Massenspektrometrie bestimmt. Gensequenzierungen identifizieren Mutationen bei hereditären Amyloidosen.
Therapie
Die Therapie richtet sich jeweils nach der auslösenden Ursache und der konkreten Manifestationsform der Amyloidose. Bei der AL-Amyloidose ist z.B. eine Chemotherapie mit anschließender Stammzelltransplantation möglich. Die AA-Amyloidose wird z.T. mit Colchicin behandelt.
Für die Behandlung einer ATTR-Amyloidose stehen verschiedene Medikamente mit unterschiedlichen Wirkmechanismen zur Verfügung:.[1][2][3]
- Inotersen: Hemmung der Transthyretinproduktion
- Patisiran: Gene Silencing des Transthyretin-Gens
- Tafamidis: Stabilisierung des Transthyretins
Unabhängig von der Ursache werden symptomatische Allgemeinmaßnahmen ergriffen, welche die Folgen des Organbefalls lindern sollen. Bei nephrotischem Syndrom werden z.B. Diuretika, Kompressionsstrümpfe und ggf. Albumininfusionen eingesetzt. ACE-Hemmer sollten nur mit Vorsicht verwendet werden. Sie zeigen keine Wirkung auf das Fortschreiten der Nephropathie.
Eine chronische Herzinsuffizienz bei Kardiomyopathie wird ebenfalls mit Diuretika behandelt. Digitalis, Calciumkanalblocker und Betablocker sind relativ kontraindiziert, da sie mit Amyloidfibrillen interagieren, einen AV-Block und eine Verschlechterung der Herzinsuffizienz hervorrufen können. Amiodaron wird bei Herzrhythmusstörungen eingesetzt. Ein implantierbarer Kardioverter-Defibrillator kann bei einigen Patienten sinnvoll sein. Bei Vorhofflimmern kommt eine Vorhofablation infrage.
Autonome Neuropathien werden z.B. mit Midodrin behandelt, gastrointestinale Dysfunktionen ggf. mit Prokinetika und Ballaststoffen.
Quellen
- ↑ EMA- Zusammenfassung des Merkmals Inotersen, abgerufen am 30.09.2021
- ↑ EMA- Zusammenfassung des Merkmals Patisiran, abgerufen am 30.09.2021
- ↑ Fachinformation Vyndaqel, abgerufen am 30.09.2021
Literatur
- Ladner-Merz S, Müller-Ladner U. 108 Amyloidose. In: Suttorp N, Möckel M, Siegmund B et al., Hrsg. Harrisons Innere Medizin. 20. Auflage. Berlin: ABW Wissenschaftsverlag; 2020.