Amyotrophe Lateralsklerose










Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
Loslegenvon altgriechisch: μῦς ("mys") - Muskel, τροφή ("trophḗ") - Nahrung
Synonyme: amyotrophische Lateralsklerose, myatrophe Lateralsklerose, Charcot-Syndrom, Lou-Gehrig-Syndrom, MAL
Englisch: amyotrophic lateral sclerosis
Definition
Die amyotrophe Lateralsklerose, kurz ALS, ist eine chronisch-degenerative Motoneuronenerkrankung. Sie verläuft progressiv und geht mit einer Atrophie der Skelettmuskulatur einher. Es werden mehrere Verlaufsformen unterschieden.
Epidemiologie
Die amyotrophe Lateralsklerose manifestiert sich üblicherweise in der zweiten Lebenshälfte und weist einen Häufigkeitsgipfel um das 7. Lebensjahrzehnt auf; Männer sind häufiger betroffen als Frauen. Die Literatur gibt eine Inzidenz von 1 - 2 Fällen auf 100.000 Personen an, die Prävalenz liegt bei etwa 5:100.000.
Für die amyotrophe Lateralsklerose sind Endemiegebiete beschrieben, beispielsweise auf Guam im Westpazifik. Die Inzidenz für die Erkrankung ist hier bis zu fünfzigfach erhöht. Dabei wird die von Cyanobakterien gebildete Aminosäure β-Methylamino-L-Alanin (BMAA) als Ursache diskutiert.[1] Diese Vermutung ist jedoch umstritten.
Ätiologie
Die Ursachen der amyotrophen Lateralsklerose sind bis heute (2026) nicht vollständig geklärt. Eine typischerweise beobachtbare familiäre Häufung von Krankheitsfällen weist auf eine genetische Disposition hin. Ein Teil der Patienten weist Mutationen im TDP-43- (TARDBP) oder SOD1-Gen auf. Weitere Mutationen betreffen die Gene FUS, TBK1 und OPTN.
Es werden aber auch andere Faktoren wie Virus- oder Autoimmunerkrankungen diskutiert. Neuere Forschungen weisen darauf hin, dass Störungen der DNA-Reparatur ein wesentlicher Auslösefaktor der amyotrophen Lateralsklerose sind.[2]
In ca. 11% aller und ca. 40% der familiären ALS-Fälle wird eine Hexanukleotidexpansion (GGGGCC) im C9orf72-Gen beobachtet. Diese Mutation findet sich auch bei einigen Fällen frontotemporaler Demenz und sehr häufig bei einem Overlap von ALS und FTD.[3][4] Die Konsequenzen dieser Expansion sind derzeit (2026) noch unklar, wahrscheinlich existieren mehrere pathogene Effekte.
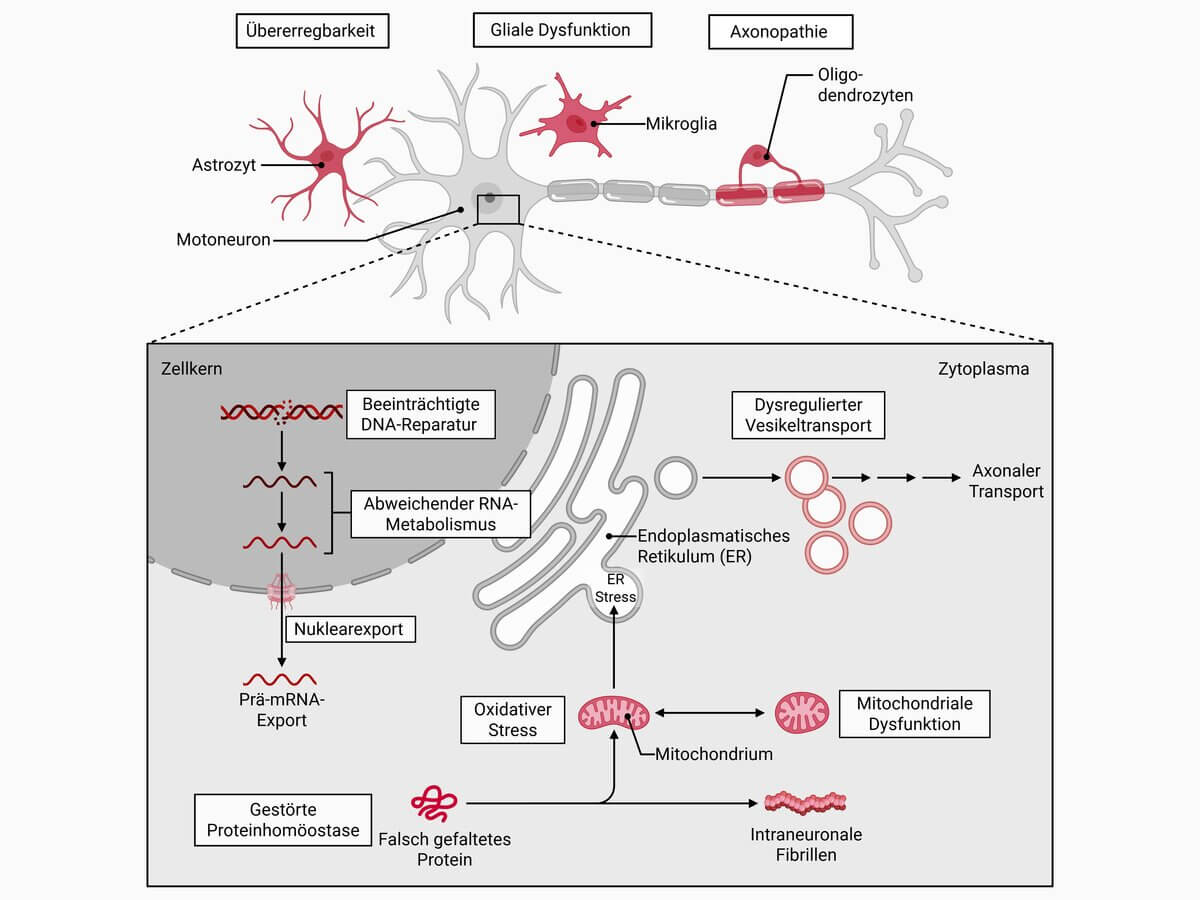
Pathophysiologie
Die Symptomatik der Erkrankung resultiert aus dem Zugrundegehen der Motoneurone des Rückenmarks, die wiederum auf eine Degeneration bestimmter Hirnareale zurückgeführt werden kann. Auch ein Schwund von Ganglienzellen in motorischen Hirnnervenkernen und dem medullären Vorderhorn lässt sich regelmäßig nachweisen. In diesen Neuronen ist das Auftreten von Bunina-Körpern typisch für eine ALS.
Durch die neuronale Degeneration kommt es sekundär zu einer Atrophie der innervierten Muskulatur, besonders an den Extremitäten; die Symptome ähneln denen einer spinalen Atrophie. Ausfälle motorischer Hirnnerven führen zur Atrophie der Gesichtsmuskulatur und zu einer progressiven Bulbärparalyse (PBP).
Auf molekularer Ebene führen bei der amyotrophen Lateralsklerose verschiedene pathologische Prozesse zur Degeneration der Motorneurone. Dazu gehören die Fehlfaltung und Aggregation der o.a. Proteine TDP-43, SOD1 und FUS, welche die Funktion von Neuronen und Gliazellen beeinträchtigen. Es kommt zu einer gestörten Proteostase, toxische Proteinaggregate werden nicht mehr ausreichend abgebaut.
Systematik
Man unterscheidet zwei Hauptformen der amyotrophen Lateralsklerose:
- Sporadische Form: Die sporadische amyotrophe Lateralsklerose tritt am häufigsten auf. Ihre Ursachen sind bis heute nicht verstanden.
- Familiäre Form: Die familiäre amyotrophe Lateralsklerose ist eine autosomal-dominante Erkrankung. Es werden mehrere genetische Subtypen unterschieden.
Die endemische Form (siehe oben) spielt nur eine untergeordnete Rolle.
Symptomatik
Die amyotrophe Lateralsklerose manifestiert sich zuerst an den Akren durch unkontrollierte Faszikulationen und später schlaffe Lähmungen. Von den kleinen Hand- und Fußmuskeln schreitet die Erkrankung nach proximal fort, wo sie die Muskelgruppen an Armen und Beinen atrophieren lässt. Neben den schlaffen Paralysen nehmen viele Patienten äußerst schmerzhafte Muskelkrämpfe wahr.
Die Atrophie der Gesichtsmuskulatur lässt das Gesicht ausdruckslos und eingefallen erscheinen. Schließlich manifestieren sich Lähmungen der Muskulatur an Zunge, Gaumen, Pharynx und Larynx (Symptomatik der Bulbärparalyse), die schließlich zum Tod durch Aspiration von Fremdkörpern oder Erstickung führen.
Sensibilität, Sensorik und Bewusstsein sind während des Krankheitsverlaufs meist erhalten. In späteren Stadien kann es jedoch zur Entwicklung einer frontotemporalen Demenz kommen.[5]
Anmerkung: Eine Sonderform der amyotrophen Lateralsklerose führt zu spastischen Lähmungen besonders der Beine, Rigidität und positiven Pyramidenbahnzeichen. Sie wird als Erb-Sklerose bezeichnet.
Diagnostik
- Körperliche Untersuchung: Der erste Schritt zur Diagnose einer amyotrophen Lateralsklerose besteht in der körperlichen bzw. neurologischen Untersuchung. Können durch Beklopfen der Muskulatur oder durch Kältereize Faszikulationen ausgelöst werden, besteht ein weiter abzuklärender Krankheitsverdacht.
- Elektromyogramm: Im Elektromyogramm lässt sich üblicherweise eine pathologische Muskelaktivität, häufig kombiniert mit einer Verminderung motorischer Einheiten, nachweisen.
- Bildgebung: Nachweis von Atrophien in Gehirn und Rückenmark durch CT oder MRT
- Labordiagnostik: Nachweis von Neurofilament light (NFL) im Liquor oder im Blut. Der NFL-Spiegel ist bei ALS signifikant erhöht. Die Höhe des Serumspiegels ermöglicht eine Prognoseabschätzung.[6][7]
- Muskelbiopsie: Sie ermöglicht die Differenzierung zwischen neurogener und myogener Muskelatrophie, ist aber keine Standarddiagnostik bei ALS.
Die Diagnose wird anhand der Gold-Coast-Kriterien oder der El-Escorial-Kriterien gestellt.
Differentialdiagnosen
Therapie
Zurzeit (2026) existiert keine kurative Therapie für die amyotrophe Lateralsklerose. Die Therapie ist multimodal und zielt auf eine Verlangsamung der Krankheitsprogression sowie eine Verbesserung der Symptomatik. Dabei sollte die Lebensqualität der Patienten im Vordergrund stehen. Die frühzeitige Einbindung einer palliativmedizinische Mitbetreuung (z.B. durch ein spezialisiertes ambulantes Palliativteam oder eine neuropalliative Sprechstunde) ist häufig sinnvoll, um Symptomkontrolle, psychosoziale Unterstützung, Advance Care Planning und Krisenpläne strukturiert zu etablieren.
Krankheitsverzögerung
Seit 2024 ist das Antisense-Oligonukleotid Tofersen zur Therapie der SOD1-assoziierten ALS bei Erwachsenen, verfügbar. Es beruht auf dem Prinzip des Gene-Silencing. Tofersen bindet an die SOD1-mRNA und fördert dadurch deren Abbau. Die SOD1-Proteinsynthese wird supprimiert. Es kommt zu einer Verringerung der Neurofilament-Leichtketten im Plasma der behandelten Patienten und damit zu einer verlangsamten Krankheitsprogression.[8]
Der seit 1996 zugelassene Glutamat-Antagonist Riluzol ermöglicht die Verzögerung des Fortschreitens der Krankheit. Ggf. kann Riluzol mit dem Anti-Parkinsonmittel Rasagilin kombiniert werden.[9] Einen starken Einfluss auf die Mortalität haben die verfügbaren Wirkstoffe nicht.
Symptomatische Therapie
Die symptomatische Therapie spielt eine wichtige Rolle. Nach der Bildung der bulbären Symptomatik ist für die meisten Patienten die Hypersalivation das größte Problem. Der Patient kann den Speichel nicht mehr richtig schlucken und verschluckt sich dauernd. Hier kommen anticholinerg wirksame Medikamente wie Amitriptylin, Scopolamin oder Methionin zum Einsatz, deren "Nebenwirkungen" man nutzt, um die Hypersalivation zu vermindern. Ersatzweise bietet sich eine Injektion von Botox in die Speicheldrüsen an.
Eine Bestrahlung der Speicheldrüsen kann versucht werden, wenn die pharmakologische Behandlung keine Erfolge erzielt.
Bei Angst und Unruhezuständen werden schnell wirksame Benzodiazepine eingesetzt. Dazu eignet sich sublingual und buccal applizierbares Lorazepam, da die Patienten im fortgeschrittenen Stadium nicht mehr schlucken können. Es können aber auch Diazepam-Tropfen über die PEG gegeben werden.
Wichtiger Bestandteil der Therapie ist die psychologische, vor allem neuropsychologische und psychotherapeutische Betreuung der Patienten, die das Fortschreiten bei vollem Bewusstsein erleben. Die Psychotherapie kann zur Stärkung des Lebenswillens beitragen und ermöglicht dem Patienten einen besseren Umgang mit der Krankheit.
Bei der bulbären Verlaufsform kann eine Logopädie Symptome wie Schluckstörungen oder Bisse in die Zunge bzw. Wangen mildern und hilft bei der Sprechstörung.
In der terminalen Krankheitsphase ist die Angst zu ersticken eines der größten Probleme. Ihr wird mit niedrig dosierten Opiaten wie Morphin begegnet. Opiate wirken anxiolytisch und unterdrücken das Atemzentrum in Medulla oblongata. Dadurch werden der Atemantrieb und die subjektive Dyspnoe reduziert.
Bei progressiven Formen der amyotrophen Lateralsklerose müssen eine invasive Unterstützung der Atmung sowie eine Sondenernährung zur Aspirationsprophylaxe mit dem Patienten diskutiert werden.
Physiotherapie
Die Physiotherapie dient der Erhaltung von Muskelkraft und Beweglichkeit. Sie umfasst aktive und passive Mobilisationsübungen, Kräftigungs- und Dehnprogramme sowie Maßnahmen zur Sturzprophylaxe. Ziel ist es, Kontrakturen zu vermeiden und die Mobilität des Patienten möglichst lange zu erhalten. Ergänzend sind Atemtherapie und Hilfsmittelberatung sinnvoll.
Stammzelltherapie
Ein weiterer Behandlungsansatz ist die Stammzelltherapie. Dabei werden autologe Stammzellen eingesetzt, die dem Patienten entweder aus dem Knochenmark oder dem Blut entnommen, gezüchtet und dann in geschädigte Areale des Großhirns und des Rückenmarks eingepflanzt werden. Eine abschließende Beurteilung ist zur Zeit (2026) nicht möglich, da die Datenlage ungenügend ist. Diese Form der Therapie ist in Deutschland nicht zugelassen.
Weitere experimentelle Ansätze
Der Arzneistoff Edaravon (3-Methyl-1-phenyl-5-pyrazolon) soll ebenso wie Riluzol zu einer leichten Verzögerung des Krankheitsprozesses führen (wenige Monate). Der Wirkmechanismus ist bisher (2026) unbekannt. Edaravon ist nur in Japan, der Schweiz und den USA zugelassen.
Die 2022 in den USA vorläufig zugelassene Kombination aus Natriumphenylbutyrat und Ursodoxicoltaurin (AMX0035, Relyvrio®) konnte in einer 2024 beendeten Phase-III-Studie keine ausreichende Wirksamkeit nachweisen.
In Tierversuchen führte das Antibiotikum Minocyclin zu einer deutlichen Lebensverlängerung erkrankter Tiere. Die Anwendung des Präparats bei ALS-Patienten zeigte jedoch in einer Studie eine destruktive und krankheitsbeschleunigende Wirkung.[10]
Der Hitzeschockprotein-Inducer Arimoclomol oder der Komplement-C5-Inhibitor Ravulizumab sind bei der Behandlung der amyotrophen Lateralsklerose nicht wirksam.[11]
Prognose
Die amyotrophe Lateralsklerose führt üblicherweise innerhalb weniger Jahre zum Tod durch eine respiratorische Insuffizienz, die zu einer Hypoxämie und Hyperkapnie führt. Durch die verminderte Lungenbelüftung treten häufig Pneumonien auf, die den Zustand des Patienten akut verschlechtern können.
Die mittlere Überlebensdauer nach Diagnosestellung beträgt 3 Jahre, es sind aber auch Fälle mit einem deutlich längeren Überleben geschildert. Das bekannteste Beispiel hierzu ist sicherlich der Astrophysiker Stephen Hawking, der seit seiner Diagnosestellung 1963 über fünfzig Jahre überlebte und erst im Jahr 2018 verstarb.
Quiz
Leitlinie
- S1-Leitlinie Motoneuronerkrankungen der Deutschen Gesellschaft für Neurologie, AWMF-Registernummer: 030/001 (Stand 2021)
- S2k-Leitlinie Palliativmedizinische Versorgung neurologischer Erkrankungen, zuletzt abgerufen am 13.03.2026
Bildquelle
- Bildquelle für Flexikon-Quiz: © Major Tom Agency / Unsplash
Quellen
- ↑ Bishop SL, Murch SJ (2020) A systematic review of analytical methods for the detection and quantification of β- N -methylamino- l -alanine (BMAA). Analyst 145:13–28.
- ↑ ALS: Dresdner Grundlagenforscher entdecken neuen Krankheitsmechanismus bei Amyotropher Lateralsklerose
- ↑ DeJesus-Hernandez et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron, 2011.
- ↑ Smeyers, Banchi, Latouche. C9ORF72: What It Is, What It Does, and Why It Matters. Frontiers in Cellular Neuroscience, 2021.
- ↑ Mattle und Fischer. Kurzlehrbuch Neurologie. 5., überarbeitete Auflage. Thieme Verlag Stuttgart, 2021.
- ↑ Ching-Hua Lu et al.: Neurofilament light chain. A prognostic biomarker in amyotrophic lateral sclerosis Neurology. 2015 Jun 2; 84(22): 2247–2257. doi: [10.1212/WNL.0000000000001642] PMCID: PMC4456658 PMID: 25934855
- ↑ Verde F, Steinacker P, Weishaupt JH, et al.: Neurofilament light chain in serum for the diagnosis of amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry Published Online First: 11 October 2018. doi: 10.1136/jnnp-2018-318704
- ↑ Full prescribing Information Qalsody, FDA, abgerufen am 27.04.2023
- ↑ Ludolph AC et al.: Safety and efficacy of rasagiline as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: a randomised, double-blind, parallel-group, placebo-controlled, phase 2 trial. Lancet Neurol. 2018 Aug;17(8):681-688. doi: 10.1016/S1474-4422(18)30176-5. Epub 2018 Jun 19.
- ↑ Gordon et al. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol; 2007
- ↑ Diener HC, Highlights der klinischen Neurologie: Neurology in Progress. Vortrag DGN Kongress 2024, Blog Pharmakotherapie, abgerufen am 14.11.2024