Hypertrophe Kardiomyopathie











Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenSynonyme: familiäre hypertrophe Kardiomyopathie, FHC
Englisch: hypertrophic cardiomyopathy, familial hypertrophic cardiomyopathy
Definition
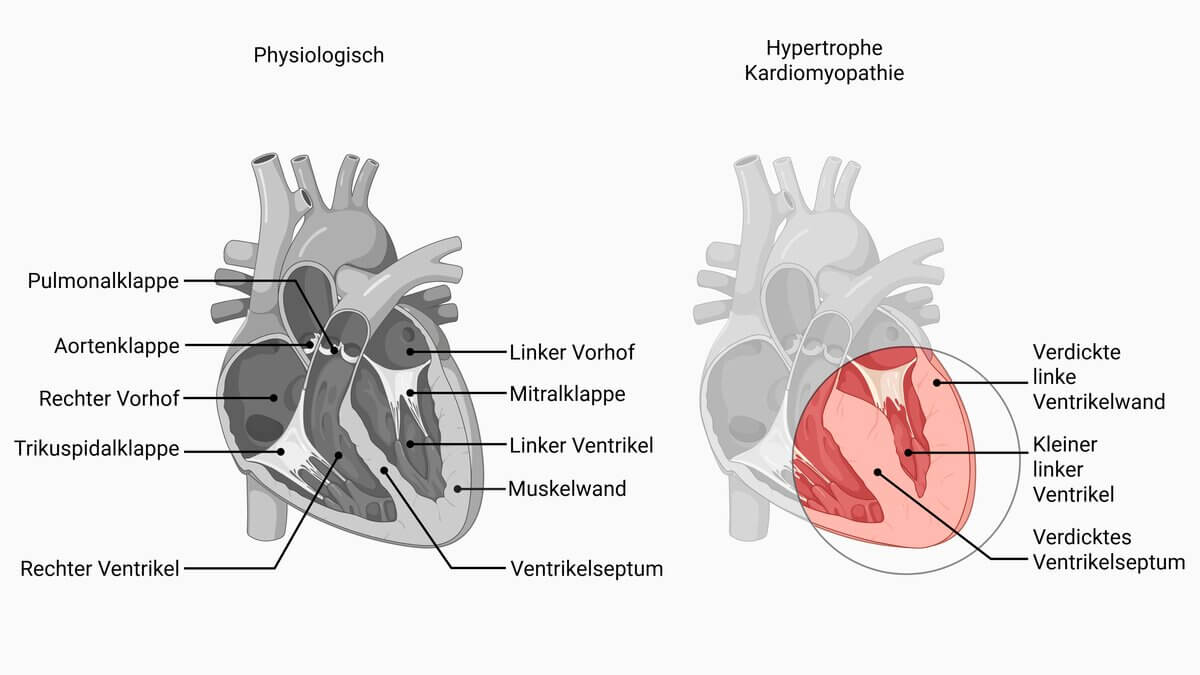
Die hypertrophe Kardiomyopathie, kurz HCM, ist eine Muskelerkrankung des Herzens (Kardiomyopathie). Sie ist durch Mutationen verschiedener myokardialer Strukturproteine gekennzeichnet und wird meist autosomal-dominant vererbt. Die HCM führt überwiegend zu einer asymmetrischen Hypertrophie des linken Ventrikels, die nicht durch Druck- oder Volumenbelastung bedingt ist.
Epidemiologie
Die HCM hat eine Prävalenz von etwa 0,2 % in der Allgemeinbevölkerung und ist damit zwar selten, aber die häufigste genetische Kardiomyopathie. Sie ist bei beiden Geschlechtern gleich häufig. Die HCM ist die häufigste Ursache des plötzlichen Herztods (SCD) bei Jugendlichen und jungen Erwachsenen, insbesondere bei Leistungssportlern. Vor dem 60. Lebensjahr sind die meisten HCM-bedingten Todesfälle plötzliche Herztode, während ältere Patienten häufiger an Herzinsuffizienz oder Schlaganfall versterben.
Ätiologie
Bei der hypertrophen Kardiomyopathie handelt es sich überwiegend um eine autosomal-dominante Erkrankung mit unvollständiger Penetranz. Autosomal-rezessive, X-chromosomale und mitochondriale Formen sind beschrieben. Es existiert eine Vielzahl an möglichen Genmutationen, insbesondere der am Aufbau des Sarkomers beteiligten Strukturproteine. Betroffene Gene sind u.a.:
- MYBPC3 (myosinbindendes Protein) auf Chromosom 11: 30 bis 40 %
- MYH7 (schwere Myosinkette) auf Chromosom 14: 20 bis 30 %
- TNNT2 (kardiales Muskeltroponin) auf Chromosom 1: 10 %
- TNNI3 (Troponin I Typ 3) auf Chromosom 19: 7 %
- MYL2 (leichte Myosinkette 2) auf Chromosom 12: 4 %
- MYL3 (leichte Myosinkette 3) auf Chromosom 3: 2 %
- TPM1 (Tropomyosin 1) auf Chromosom 15: 1 %
Auch ein syndromales Auftreten, z.B. beim Noonan-Syndrom, bei der Friedreich-Ataxie oder im Rahmen einer Embryopathia diabetica ist möglich. Bei Erstdiagnose wird eine genetische Testung mittels Sarkomer-Gen-Panel empfohlen, um weitere Mutationsträger in der Familie zu identifizieren.
Pathophysiologie
Bei der hypertrophen Kardiomyopathie kommt es zu einer Verdickung der Muskulatur des linken Ventrikels (> 12 bis 15 mm diastolisch) ohne begleitende Dilatation. In 15 bis 17 % findet sich eine assoziierte rechtsventrikuläre Hypertrophie.
In 70 % der Fälle liegt eine Obstruktion des linksventrikulären Ausflusstraktes (LVOT) vor. Man spricht dann von einer hypertroph-obstruktiven Kardiomyopathie (HOCM). Sie ist definiert durch einen LVOT-Gradienten > 30 mmHg, wobei man ab 50 mmHg von einer hämodynamischen Relevanz ausgeht. Die Obstruktion betrifft meist das basale Septum.
Weiterhin ist die HCM mit einem "Systolic Anterior Movement" (SAM) des anterioren und seltener des posterioren Mitralklappensegels assoziiert. Ein SAM kann die LVOT-Obstruktion verstärken.
Weitere assoziierte Begleitbefunde sind:
- Mitralklappeninsuffizienz: klassischerweise nach posterior gerichtet
- linksatriale Dilatation
- Pathologien der Papillarmuskeln: z.B. Hypertrophie oder direkter Ansatz am anterioren Segel der Mitralklappe.
Pathohistologie
Die pathohistologischen Veränderungen der HCM sind charakteristisch, jedoch nicht vollständig spezifisch. Typische Befunde sind:
- ausgeprägte Hypertrophie der Kardiomyozyten mit vergrößerten, hyperchromatischen Zellkernen
- Desorganisation der Myokardfasern (myocyte disarray) mit ungeordneter Ausrichtung der Kardiomyozyten und Myofibrillen
- interstitielle und perivaskuläre Fibrose unterschiedlichen Ausmaßes
- Verdickung und Lumeneinengung kleiner intramuraler Koronararterien (small vessel disease)
Das Ausmaß der Fibrose korreliert mit dem Risiko für ventrikuläre Arrhythmien und plötzlichen Herztod. Histologisch nachweisbare Fibroseareale entsprechen häufig Regionen eines Late Gadolinium Enhancement (LGE) in der Kardio-MRT.
Einteilung
...nach Hämodynamik
- HCM ohne dynamische Obstruktion (auch: hypertrophe nicht-obstruktive Kardiomyopathie, HNCM)
- HCM mit dynamischer Obstruktion (auch: hypertroph-obstruktive Kardiomyopathie, HOCM)
- Endstadium der HCM ("burned-out phase"): systolische Dysfunktion mit linksventrikulärer Ejektionsfraktion von < 50 %
...nach Morphologie
Asymmetrische HCM
Die mit 60 bis 70 % häufigste Form ist die asymmetrische HCM, bei der meist die basalen anteroseptalen und angrenzenden basalen anterioren Segmente hypertrophiert sind. Die daraus resultierende Verengung des LVOT kann zu einer dynamischen Obstruktion führen. Charakteristisch sind eine Septumdicke von ≥ 15 mm und ein Verhältnis des Septums zur inferolateralen Ventrikelwand von > 1,3 bis 1,5.
Konzentrische HCM
Die zweithäufigste Form ist die konzentrische HCM, bei der es zu einer diffusen linksventrikulären Hypertrophie kommt. Dabei müssen eine reaktive Hypertrophie aufgrund einer erhöhten Nachlast (arterielle Hypertonie, Aortenklappenstenose) und eine physiologische Hypertrophie (z.B. Sportlerherz) ausgeschlossen werden.
Apikale HCM
Die apikale hypertrophe Kardiomyopathie wird auch als Yamaguchi-Syndrom bezeichnet. Dabei sind vorwiegend die apikalen Segmente betroffen, isoliert oder in Kombination mit einer Septumhypertrophie. Hinweisend ist eine apikale Wanddicke von ≥ 15 mm und ein Verhältnis von apikaler zu basaler Wanddicke von > 1,5. Die apikale Hypertrophie erinnert morphologisch an einen Spaten ("spade-like deformity").
Weitere Formen
- Midventrikuläre HCM: macht ca. 10 % der Fälle aus. Sie betrifft vor allem die Segmente in der Mitte des Ventrikels, was zu einer "Sanduhr"- oder "Hantel"-Form führt.
- tumefaktive HCM: isolierte fokale Verdickung eines Myokardsegments
Symptome
Die Patienten leiden unter den Symptomen einer Linksherzinsuffizienz. Typische Beschwerden sind:
- Leistungsminderung
- Müdigkeit
- Dyspnoe
- Angina pectoris-Anfälle
- ventrikuläre Arrhythmien (bis hin zu ventrikulären Tachykardien)
- Schwindel
- Synkopen
Diagnostik
Die HCM wird klinisch diagnostiziert, wobei bildgebende Befunde hinweisend sind und den Ausschluss von Differenzialdiagnosen ermöglichen. Das EKG ist in 95 % der Fälle pathologisch:
- hohe präkordiale QRS-Amplituden, ggf. erfüllte Sokolow-Lyon-Kriterien
- sekundäre Repolarisationsstörungen: ST-Senkungen, T-Inversion
- Abweichung der Herzachse nach links
- tiefe schmale Q-Zacke in I, aVL, V5 und V6
Klinische Untersuchung
Bei der HOCM ist auskultatorisch ein hartes, rautenförmiges Systolikum mit punctum maximum über dem 4. Interkostalraum links parasternal typisch. Charakteristisch ist die Verstärkung durch Provokationsmanöver, die die Vorlast senken – wie das Valsalva-Manöver oder das Aufstehen aus der Hocke; eine Abschwächung erfolgt durch isometrische Belastung oder Hocken. Dieses dynamische Verhalten unterscheidet das HCM-Geräusch von anderen Ursachen einer LVOT-Obstruktion.
Bildgebung
Röntgen-Thorax
Der Röntgenbefund kann normal sein oder unspezifische Merkmale aufweisen, z.B. eine Kardiomegalie oder eine pulmonalvenöse Stauung. Die linksventrikuläre Hypertrophie ist bei der HCM nach innen gerichtet ist und das Ventrikellumen verkleinert. Im Röntgen-Thorax sieht man deshalb häufig keine erkennbare Vergrößerung der Herzsilhouette. Bei fortgeschrittener Erkrankung können Zeichen einer Herzinsuffizienz auftreten, z.B. im Sinne einer pulmonalen Stauung oder von Pleuraergüssen.
Echokardiographie
Die Echokardiographie wird zur Beurteilung der LVOT-Obstruktion, zur Quantifizierung der systolischen und diastolischen linksventrikulären Funktion, zur Erkennung einer systolischen Vorwärtsbewegung der Mitralklappe (SAM) und zur Messung der linksatrialen Größe eingesetzt. Typische Befunde in der 2D-Bildgebung sind:
- Verhältnis zwischen septaler und inferolateraler Wanddicke > 1,3 bzw. > 1,5 bei arterieller Hypertonie
- Myokardsegment mit einer Dicke > 15 mm ohne andere erklärbare Ursache
Im M-Mode kann ein Systolic Anterior Movement (SAM) auffallen: Das anteriore Mitralklappensegel wird während der Ventrikelsystole in den LVOT in Richtung Septum verschoben. In schweren Fällen kommt das Segel in der Midsystole mit dem Septum in Kontakt. Die Folge ist eine Einkerbung der M-Mode-Kurve. Bei Vorliegen eines SAM der Mitralklappe kann im Farbdoppler ein nach posterior gerichteter Jet die Mitralregurgitation anzeigen. Bei Vorliegen einer Obstruktion fällt weiterhin ein Aliasing im LVOT auf.
Für die Beurteilung der Art, des Schweregrads und der anatomischen Lage der Obstruktion kommen der Pulsed-wave Doppler (PWD) und der Continuous Wave Doppler (CW-Doppler) zum Einsatz.
Magnetresonanztomographie
Die Kardio-MRT dient der Beurteilung der Herzmorphologie und -funktion. Ihre Darstellung der anterolateralen und apikalen Segmente ist der Echokardiographie überlegen. Typische Befunde sind:
- linksventrikuläre systolische Dysfunktion
- linksventrikuläre Hypertrophie: insbesondere des basalen Septums
- SAM der Mitralklappe: mit Regurgitationsjet
- Herzspitzenaneurysma
- Late Gadolinium Enhancement (LGE) vom Nicht-Infarkt-Typ: an der rechtsventrikulären Insertion bzw. im Bereich der maximalen Hypertrophie – dabei subepikardial und intramyokardial und fleckig bis flächig
Die MRT dient weiterhin der Abgrenzung von anderen Ursachen einer linksventrikulären Hypertrophie, wie Morbus Fabry oder kardiale Amyloidose. Zudem kann sie helfen, eine tumefaktive HCM von einem Herztumor abzugrenzen. Die kardiale MRT spielt auch bei asymptomatischen Mutationsträgern eine Rolle, da sie phänotypische Marker für eine subklinische HCM bei fehlender linksventrikulärer Hypertrophie identifiziert. Dazu zählen:
- myokardiale Krypten
- elongierte Mitralklappensegel
- fleckiges bzw. streifiges LGE bzw. Fibrose in der rechtsventrikulären Insertionsstelle
Differentialdiagnosen
- Linksherzhypertrophie durch arterielle Hypertonie: häufigste Ursache einer konzentrischen linksventrikulären Hypertrophie. Kein oder minimales LGE.
- Linksherzhypertrophie durch Aortenstenose: eingeschränkte Klappenbewegung mit erhöhtem transvalvulärem Gradienten. Strukturelle Anomalie der Aortenklappe. Meist konzentrische Hypertrophie.
- Sportlerherz: konzentrische Hypertrophie mit einer Wanddicke von normalerweise 13 bis 15 mm. Adäquate linksventrikuläre Dilatation < 6,5 cm. Normales Verhältnis von Ventrikeldiameter und Myokarddicke. Normale Ejektionsfraktion und diastolische Funktion. Kein LGE. Reversibilität der Hypertrophie (nach Dekonditionierung von 9–12 Wochen).
- Danon-Erkrankung: Skelettmuskelschwäche und geringe mentale Retardierung. Mid-myokardiales LGE. Das Septum ist meist ausgespart.
- Infiltrative Kardiomyopathien:
- Morbus Fabry: LGE basal inferolateral in mid-myokardialer oder subepikardialer Lokalisation.
- Hämochromatose: Eisenablagerung ist in T2*-Sequenzen erkennbar.
- Amyloidose (meist AL- und ATTR-Amyloidose): diffuses, subendokardiales LGE. Hohes Signal in T1-Mapping. Apical Sparing (Reduktion der myokardialen Muskelverkürzung bei Kontraktion mit Aussparung des Apex). Echoreiches, septales Myokard (Granular Sparkling). Verdickung der Herzklappen und des Vorhofseptums. Konzentrische Hypertrophie. Spezifische Diagnostik mittels Skelettszintigraphie, Bestimmung der freien Leichtketten, Proteinelektrophorese und Immunfixation im Serum/Urin und ggf. Knochenmarkpunktion.
- Glykogenspeicherkrankheiten
- Dynamische LVOT-Obstruktion bei
- Hypovolämie
- distributivem Schock
- Myokardischämie im RIVA-Territorium
- sigmoidalem Kammerseptum
- Takotsubo-Kardiomyopathie
- rechtsventrikulärer Dilatation
Therapie
Die Therapie richtet sich nach dem klinischen Schweregrad. Asymptomatische Patienten mit geringem Risiko benötigen möglicherweise keine spezifische Therapie. Wettkampfsport sollte nach individueller Risikobeurteilung im Rahmen eines Shared-Decision-Prozesses in der Regel vermieden werden; Freizeitsport ist nach individueller Einschätzung häufig möglich. Bei symptomatischen Patienten ist eine medikamentöse Therapie indiziert, wobei Vasodilatatoren (u.a. ACE-Hemmer, Sartane, Nitrate, Dihydropyridine), hohe Diuretikadosen und positiv inotrope Substanzen bei bestehender LVOT-Obstruktion kontraindiziert sind bzw. nur sehr zurückhaltend eingesetzt werden sollten. Medikament der ersten Wahl sind nicht-vasodilatierende Betablocker. Alternativ kommen Calciumantagonisten wie Verapamil in Frage. Bei persistierender LVOT-Obstruktion unter Erstlinientherapie kann ergänzend Disopyramid eingesetzt werden. In ausgewählten Fällen kommt der kardiale Myosininhibitor Mavacamten zum Einsatz.
Bei begleitendem Vorhofflimmern besteht unabhängig vom CHA₂DS₂-VASc-Score eine Indikation zur Antikoagulation. Patienten mit ventrikulären Herzrhythmusstörungen können mit Antiarrhythmika oder einem implantierbaren Kardioverter-Defibrillator (ICD) behandelt werden; die ICD-Indikation wird anhand des HCM Risk-SCD Scores der ESC individuell abgeschätzt. Aufgrund des autosomal-dominanten Erbgangs wird ein kaskadenförmiges Familienscreening empfohlen.[1]
In therapierefraktären Fällen werden interventionelle und chirurgische Verfahren angewendet:
Prognose
Viele Patienten mit HCM haben keine oder nur geringe Beeinträchtigungen der Herzfunktion und daher eine normale Lebenserwartung. Besteht eine Obstruktion des linksventrikulären Ausflusstrakts, entwickelt sich eine progrediente Herzinsuffizienz. Vor allem Kinder können bei dieser Erkrankung durch einen plötzlichen Herztod sterben. Ursächlich sind ventrikuläre Herzrhythmusstörungen wie Kammerflimmern. Die jährliche Mortalität bei HCM-Erkrankten liegt bei etwa 0,5 bis 1,5 %.[2]
Negative prognostische Marker sind:
- frühere Episoden von Herzrhythmusstörungen mit oder ohne Synkope oder Herzstillstand
- SCD in der Familienanamnese
- Echokardiographie: schwere linksventrikuläre diastolische Dysfunktion, erhöhter LVOT-Spitzengradient > 80 mmHg, schwere linksventrikuläre Hypertrophie
- Kardio-MRT: LGE > 15 % des Myokards, linksventrikuläre Wanddicke ≥ 30 mm, linksventrikuläres Herzspitzenaneurysma, verminderte linksventrikuläre Ejektionsfraktion < 50 % (burned-out)
Quellen
- ↑ Arbelo E et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023;44(37):3503–3626.
- ↑ Möbius-Winkler MN, Laufs U, Lenk K. Diagnostik und Therapie der hypertrophen Kardiomyopathie. Dtsch Arztebl Int. 2024.
Literatur
- Ommen SR et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR Guideline for the Management of Hypertrophic Cardiomyopathy. Circulation. 2024;149(23):e1239–e1311.
- Amano Y et al. Cardiac MR Imaging of Hypertrophic Cardiomyopathy: Techniques, Findings, and Clinical Relevance. Magn Reson Med Sci. 2018
- Maron MS et al. How to Image Hypertrophic Cardiomyopathy. Circ Cardiovasc Imaging. 2017
- Baxi AJ et al. Hypertrophic Cardiomyopathy from A to Z: Genetics, Pathophysiology, Imaging, and Management. Radiographics. 2016
- Noureldin RA et al. The diagnosis of hypertrophic cardiomyopathy by cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2012
- Soler R et al. Magnetic resonance imaging of delayed enhancement in hypertrophic cardiomyopathy: relationship with left ventricular perfusion and contractile function. J Comput Assist Tomogr. 2006
- Hughes SE. The pathology of hypertrophic cardiomyopathy. Histopathology. 2004
- Moon JC et al. Toward clinical risk assessment in hypertrophic cardiomyopathy with gadolinium cardiovascular magnetic resonance. J Am Coll Cardiol. 2003