Mitochondriale Vererbung






Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenSynonym: mitochondrialer Erbgang
Englisch: mitochondrial inheritance
Definition
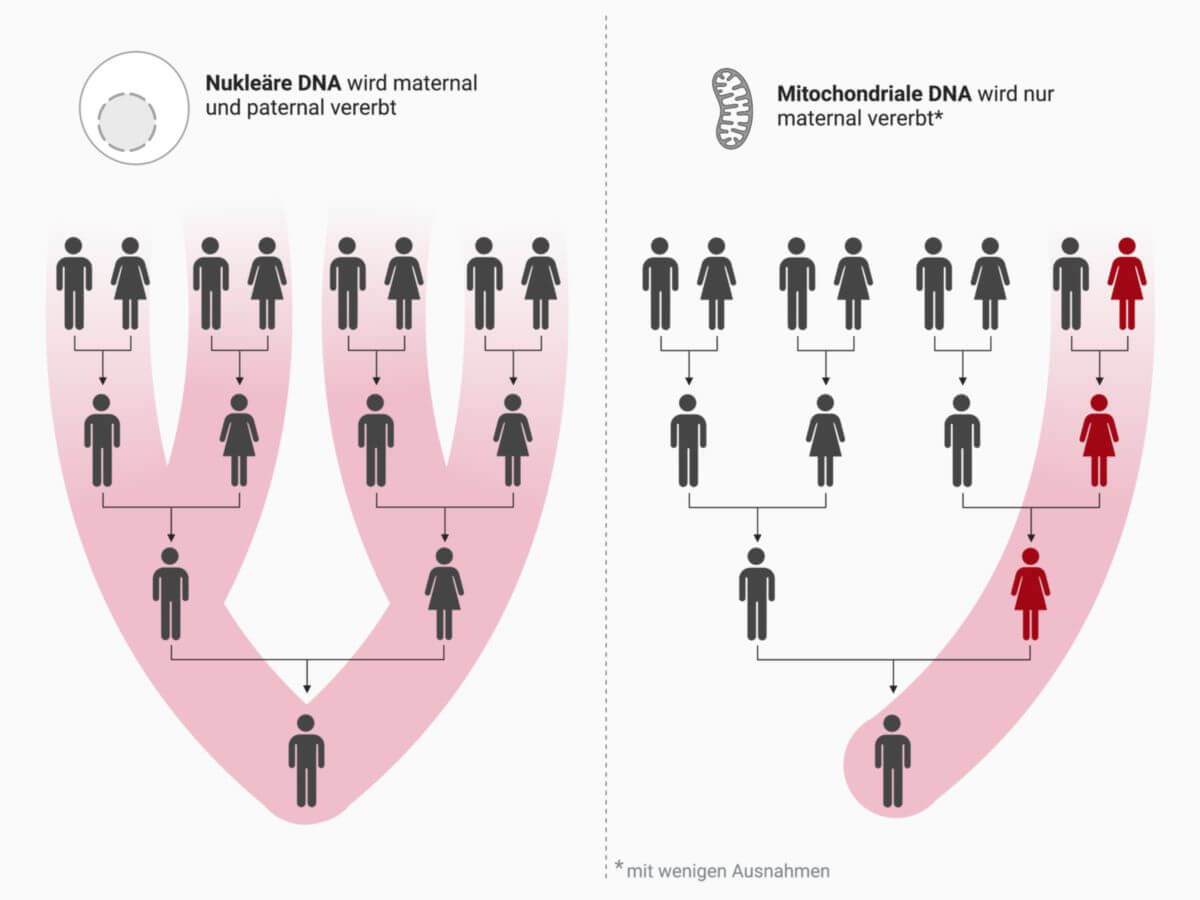
Die mitochondriale Vererbung ist eine extrachromosomale Vererbung, d.h. die betroffenen Gene befinden sich nicht auf den Chromosomen im Zellkern, sondern in den Mitochondrien. Nach dem gegenwärtigen (2025) Stand der Wissenschaft wird mitochondriale DNA (mtDNA) beim Menschen rein maternal vererbt.
Hintergrund
Das mitochondriale Genom
Kodierende DNA findet man in eukaryotischen Zellen nicht nur im nukleären Genom, sondern auch in den Mitochondrien. Man spricht von einem mitochondrialen Genom, oder mitochondrialer DNA (mtDNA). Sie liegt als ringförmiger Doppelstrang mit einer Größe von etwa 16,5 kb (Kilobasen) in 2-10 Kopien pro Mitochondrium vor. Der Großteil der eukaryontischen Zellen enthalten mehr als 1.000 mtDNA-Moleküle, die sich auf hunderte von Mitochondrien verteilen. Die reife Eizelle enthält oft mehr als 100.000 Kopien der mtDNA - damit macht die sie bis zu einem Drittel der Gesamt-DNA einer reifen Eizelle aus.
Das mtDNA-Molekül enthält insgesamt 37 Gene, von denen 13 für Proteine der Atmungskette kodieren (oxidative Phosphorylierung). Die übrigen 24 Gene kodieren 22 tRNA und 2 rRNA-Moleküle, die für die Proteinbiosynthese der 13 mitochondrial kodierten Polypeptide notwendig sind.
Alle anderen mitochondrialen Proteine sind hingegen nukleär kodiert und werden nach zytosolischer Synthese in die Mitochondrien transportiert. Der Großteil der 74 Proteine der Atmungskette (74 von 87) werden vom nukleären Genom kodiert. Dies ist auch der Grund, weshalb die meisten mitochondrialen Krankheiten auf Mutationen des nukleären Genoms beruhen.
Eine weitere Besonderheit des mitochondrialen Genoms ist, dass es nach der Vervielfältigung zufällig auf neu gebildete Mitochondrien verteilt und diese wiederum bei der Zellteilung zufällig auf die entstehenden Tochterzellen aufgeteilt werden. Ein streng kontrollierter Verteilungsmechanismus – wie er für das nukleäre Genom vorhanden ist – existiert für die mtDNA nicht. Ihre Replikation ist zudem vom Zellzyklus unabhängig.
Vererbungsvorgang
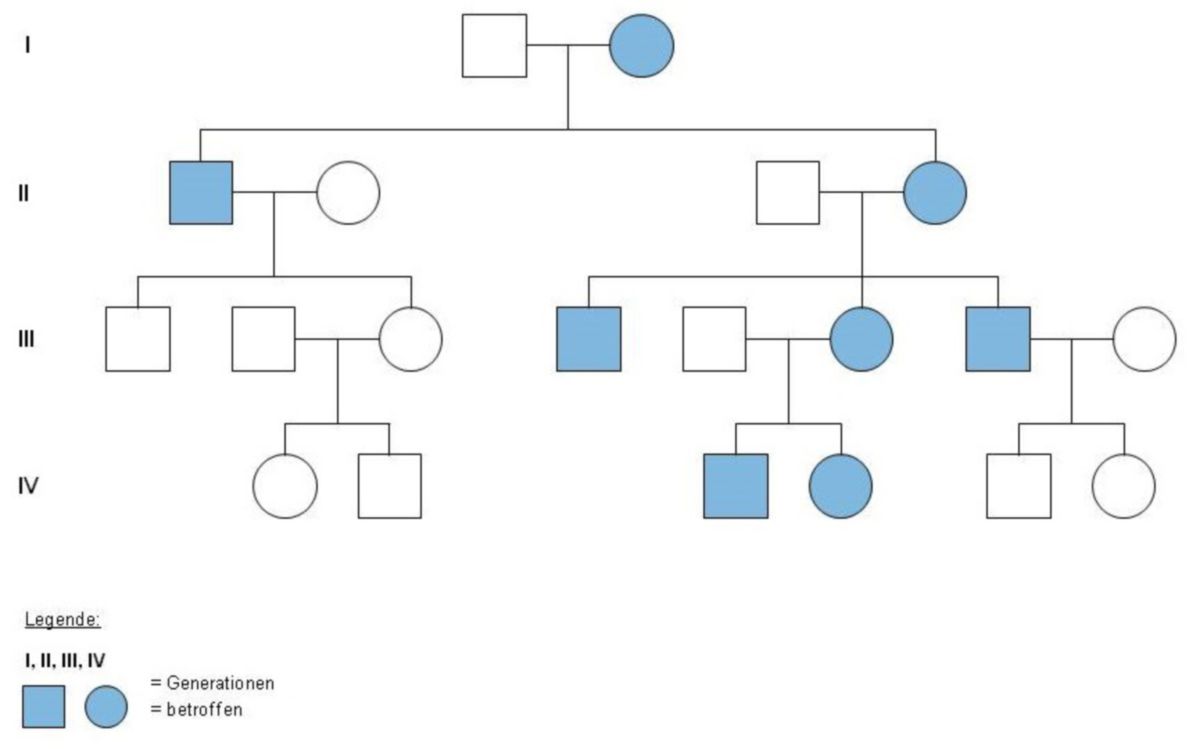
Die menschliche Zygote erhält alle ihrer Mitochondrien von der Eizelle. Grund dafür ist, dass bei der Befruchtung nur der Kopf der Samenzelle ohne Mitochondrien in die Eizelle gelangt. Mutationen der mtDNA werden von der Mutter an alle Nachkommen weitergeben.
Klinik
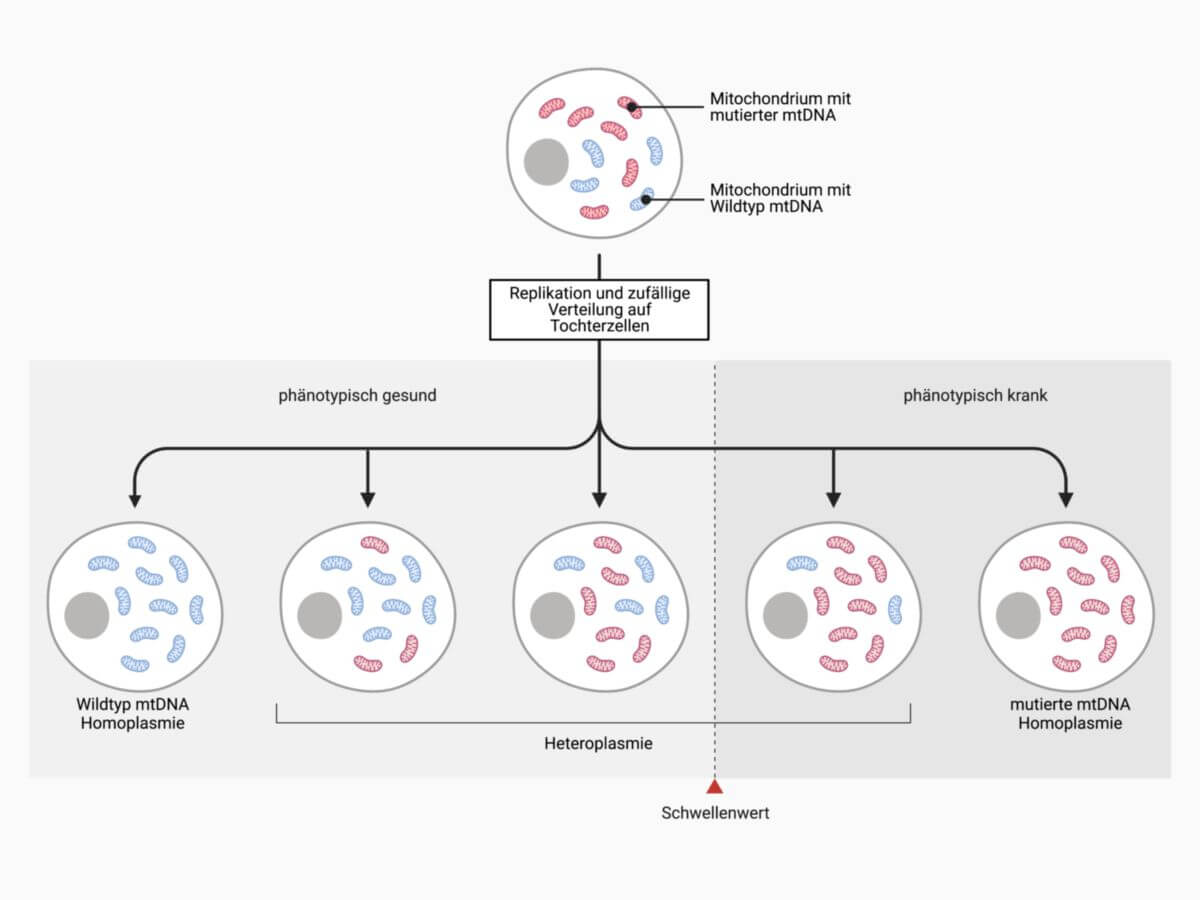
Mitochondriale Mutationen liegen auch bei symptomatischen Patienten oft nur in einem Teil der Mitochondrien einer Zelle vor. Diese Tatsache bezeichnet man als Heteroplasmie. Liegen hingegen Zellen vor, die entweder nur Mitochondrien mit normaler oder mit mutierter mtDNA enthalten, spricht man von einer Homoplasmie.
Ob sich eine mitochondriale Mutation tatsächlich phäntotypisch auswirkt, hängt vom Verhältnis zwischen normaler und mutierter mtDNA in den Zellen ab. Deshalb sind eine unvollständige Penetranz, variable Expressivität und Pleiotropie typische Merkmale für alle mitochondrial erblichen Krankheiten.
Das Verhältnis zwischen normalen und mutierten mtDNA-Kopien kann nicht nur zwischen unterschiedlichen Organen, sondern auch im Verlauf von mehreren Zellteilungen stark variieren. Es ist daher typisch, dass Patienten mit mitochondrial erblichen Krankheiten eine häufig wechselnde Symptomatik beschreiben.
Mitochondriale Mutationen können entweder direkt die Untereinheiten der Atmungskette oder aber tRNA- oder rRNA-Gene betreffen. Mutationen der RNAs wirken sich auf die Effizienz der Synthese aller mitochondrial kodierten Proteine aus – und somit ebenfalls auf die Atmungskettenfunktion.
Da mitochondriale Mutationen immer den Energiestoffwechsel der Zelle betreffen, machen sie sich besonders in stark energieverbrauchenden Geweben bemerkbar. Betroffene Organe sind somit das ZNS, die Skelettmuskulatur, die Herzmuskulatur sowie die Leber und die Nieren. Zu den typischen Krankheitsbildern zählen Enzephalopathien, Myopathien und Kardiomyopathien. Die Betroffenen können schon bei leichter Belastung den Energiebedarf nicht mehr über die oxidative Phosphorylierung decken. Folglich geraten sie zunehmend in eine Laktatazidose. Außerdem finden sich in der Skelettmuskulatur mikroskopische sogenannte ragged red fibres - subsarkolemmale Ansammlungen von Mitochondrien.
Neuromuskuläre Syndrome, denen eine Funktionsstörung der mitochondrialen Atmungskette zu Grunde liegen, werden Mitochondriopathien genannt. Beispiele für mitochondrial vererbbare Krankheiten sind:
- MELAS-Syndrom: MELAS, als Akronym verwendet, steht für Myopathie, Enzephalopathie, LaktatAzidose und Schlaganfallähnliche Episoden. Es wird typischerweise durch die Mutation 3243A>G im mitochondrialen tRNALeu-Gen verursacht.
- MERRF-Syndrom: Das MERRF steht für Myoklonische Epilepsie mit Ragged Red Fibres. Es wird typischerweise durch die Mutation 8344G>A im mitochondrialen tRNALys-Gen verursacht.
- NARP-Syndrom: Hier stehen die Buchstaben für: Neuropathie, Ataxie und Retinitis pigmentosa. Das NARP-Syndrom wird meist durch die Mutation 8993T>G oder T>C im ATPase-Gen verursacht.
- Leber-Optikusatrophie: LHON (Leber hereditäre optische Neuropathie) stellt eine Ausnahme unter den mitochondrial erblichen Krankheiten dar. Hierbei handelt es sich nicht um eine Multisystem-Krankheit. Es ist ausschließlich der Sehnerv betroffen. Häufige Ursache hierfür ist die Mutation 11778G>A in der mitochondrialen DNA.
Literatur
- Schaaf CP, Zschocke J: Basiswissen Humangenetik. Springer-Verlag
- Murken J et al.: Taschenlehrbuch Humangenetik. 8. Auflage, 2011. DOI: 10.1055/b-0034-24067
Quellen
- ↑ S. Luo, C.A. Valencia, J. Zhang, N. Lee, J. Slone, B. Gui, X. Wang, Z. Li, S. Dell, J. Brown, S.M. Chen, Y. Chien, W. Hwu, P. Fan, L. Wong, P.S. Atwal, T. Huang, Biparental Inheritance of Mitochondrial DNA in Humans, Proc. Natl. Acad. Sci. U.S.A. (2018)
- ↑ S. Lutz-Bonengel, W. Parson, No further evidence for paternal leakage of mitochondrial DNA in humans yet, Proc. Natl. Acad. Sci. U.S.A. (2019)
- ↑ Sabine Lutz-Bonengel, Harald Niederstätter et al.: Evidence for multi-copy Mega-NUMTs in the human genome, Nucleic Acids Research, Volume 49, Issue 3, 22 February 2021, Pages 1517–1531