Hepatoenzephalopathie (Hund)





Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenSynonyme: Hepatische Enzephalopathie, HE
Englisch: hepatic encephalopathy
Definition
Als Hepatoenzephalopathie des Hundes, kurz HE, bezeichnet man eine Dysfunktion des Gehirns, die sekundär bei einer Leberinsuffizienz auftritt.
Verlaufsformen
Die Erkrankung kann in unterschiedlichen Schweregraden auftreten, sodass die Symptome von mild bis zu massiv variieren können. Beim Hund wurde jedoch bisher (2022) kein Scoring-System validiert. Man unterscheidet grob zwei Formen einer Hepatoenzephalopathie:
- perakute schwere Leberdysfunktion (fulminante Hepatitis)
- chronische Form, die subklinisch bis hochgradig verlaufen kann
Ätiologie
Fulminante Hepatitis
Bei einer fulminanten Hepatitis kommt es zur akuten Nekrose des Lebergewebes. Sie wird durch Infektionen (z.B. canines Adenovirus 1) oder auch Toxine (z.B. Paracetamol, Xylitol, Pilztoxine u.ä.) verursacht. Dabei treten neben einer schweren Hepatoenzephalopathie (hepatisches Koma), Ikterus, Erbrechen und spontane Blutungsneigung aufgrund einer DIC (disseminierte intravasale Koagulopathie) auf. Die Leberenzyme sind stark erhöht und betroffene Tiere versterben binnen weniger Tage.
Portosystemischer Shunt
Die weitaus häufigere, chronische Hepatoenzephalopathie wird dadurch verursacht, dass das Blut aus der Vena portae (Pfortader) an der Leber vorbeigeleitet wird (portosystemische kollaterale Zirkulation). Der portosystemische Shunt kann sowohl angeboren als auch erworben sein. Die erworbene Form wird dabei immer durch eine Pfortaderhypertonie verursacht und deshalb auch als portosystemische Enzephalopathie bezeichnet.
Durch die besonders große Reservekapazität der Leber werden die Patienten auch bei schweren Lebererkrankungen (mit Ausnahme der fulminanten Hepatitis) vor einer Hepatoenzephalopathie geschützt. Daher reicht ein portosystemischer Shunt alleine meist nicht aus, um eine Hepatoenzephalopathie hervorzurufen. Sie entwickelt sich häufig erst in Kombination mit einer verminderten Leberfunktion, z.B. bei chronischer Hepatitis. Hier kommt es zu einer Pfortaderhypertonie und Ausbildung von portosystemischen Kollateralen.
Bei kongenitalen Shunts wird die Leberfunktion zunehmend inadäquat, da aufgrund eines Mangels an Wachstumsfaktoren das Leberwachstum hinter dem Körperwachstum zurückbleibt. Aus diesem Grund weisen Tiere mit kongenitalen Shunts eine verkleinerte Leber auf, wobei die ersten Symptome erst im Alter von ca. 6 Monaten (oder älter) auftreten.
Pathogenese
Die Pathogenese ist multifaktoriell und daher komplex.
Verantwortlich für die chronische Hepatoenzephalopathie ist eine Dysfunktion verschiedener Transmittersysteme. Zu den wichtigsten betroffenen Neurotransmittersystemen zählen der Glutamat-Dopamin/Noradrenalin-Pfad sowie der GABA/Benzodiazepin-Pfad. Das Gehirn benötigt für die Herstellung und Homöostase dieser Transmitter Vorläufer, die aus dem Darmtrakt stammen und in der Leber umgewandelt werden müssen. Aufgrund der eingeschränkten Leberfunktion haben diese Vorläufer bei einem portosystemischen Shunt unregulierten Zugang zum Gehirn.
Glutamat zählt zu den wichtigsten exzitatorischen Neurotransmittern und wird aus Ammoniak gebildet. Dieses wiederum stammt vorwiegend aus dem Darm, wo es im Kolon im Zuge des bakteriellen Abbaus von stickstoffhaltigen Substanzen (Proteinen, Amine, Harnstoff) und zusätzlich über einen Intermediärstoffwechsel in der Mukosa des Darmtrakts gebildet wird. Bei diesen Vorgängen wird Ammoniak aus Glutamin freigesetzt. Da die Leber äußerst effizient das Ammoniak aus dem Pfortaderblut entfernt (enterohepatischer Kreislauf), wird bei der Leberpassage nahezu das gesamte Ammoniak aus dem Blut gefiltert. Das Ammoniak wird dabei großteils in den Hepatozyten in Harnstoff umgewandelt und schließlich über das Blut in die Nieren transportiert, wo es über den Harn ausgeschieden wird. Beim portosystemischen Shunt fehlt diese Ammoniakumwandlung, sodass die Konzentration im Plasma ansteigt.
Pathophysiologie
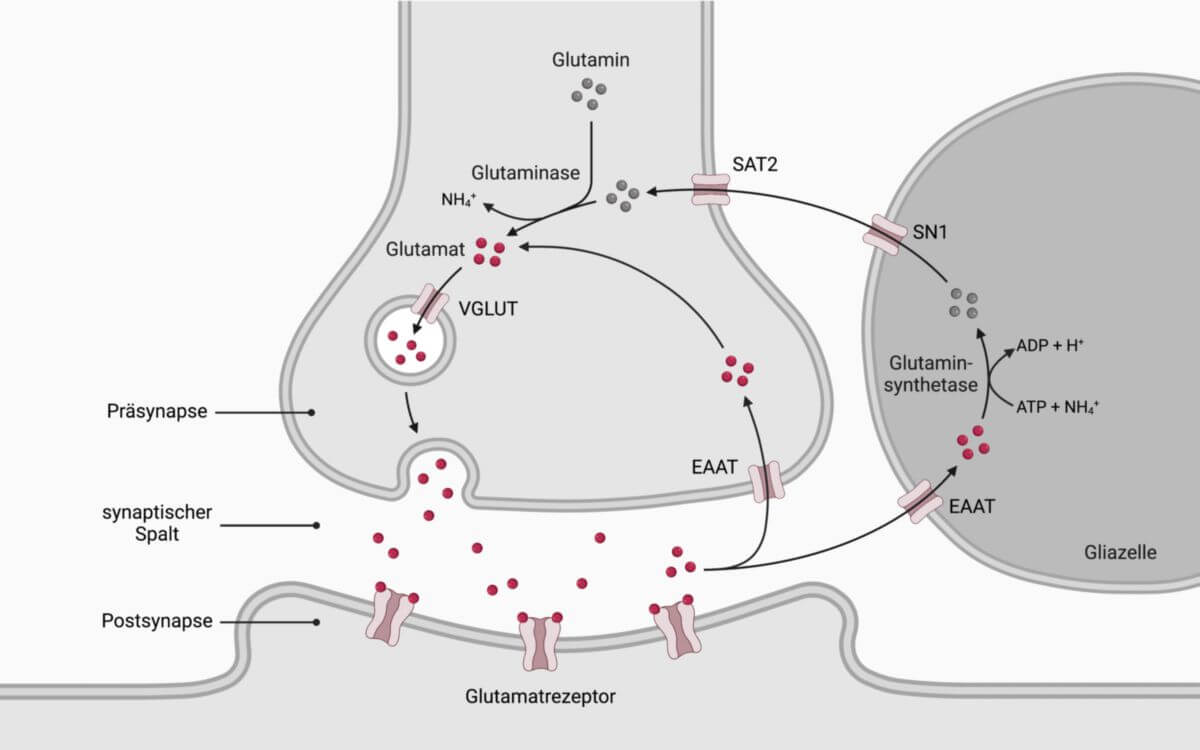
Glutamat-Glutamin-Ammoniaktransport
Ammoniak dringt in die Astrozyten ein und wird dort zur Synthese von Glutamin verwendet. Dieser Prozess ist ATP-gesteuert und wird durch die Glutaminsynthetase katalysiert. Das Glutamin diffundiert in die angrenzenden Neuronen, wo es durch die Glutaminase zu Glutamat umgewandelt wird. In den Neuronen erfolgt dann zum Teil die weitere Umwandlung in GABA. Das exzitatorische Glutamat und das inhibitorische GABA sind dabei fein aufeinander abgestimmt und regulieren so die Erregbarkeit der postsynaptischen Neuronen.
Bei einer Hyperammonämie ist die Astrozytenkapazität jedoch überlastet. Dies führt dazu, dass freies Ammoniak direkt in die Neuronen diffundiert und dort die Glutaminaseaktivität hemmt. Entsprechend kommt es zur Akkumulation von Glutamin und einem gleichzeitigen Mangel an Glutamat. Der gestörte Glutamat-Glutamin-Ammoniaktransport zwischen den Astrozyten und Neuronen ist ausschlaggebend für die Pathogenese der Hepatoenzephalopathie.
Alkalose
Die Zellmembran ist nur für die nicht-ionisierte Form des Ammoniaks (NH3) durchlässig, Ammoniumionen (NH4+) können die Membran nicht passieren. Im gelösten Zustand besteht ein Gleichgewicht zwischen beiden Formen, das vom pH-Wert abhängig ist. Im Falle einer Alkalose ist es in Richtung des NH3 verschoben, hohe pH-Werte führen somit zu einem schwereren Verlauf der Hepatoenzephalopathie.
Ausgelöst wird die Alkalose u.a. durch eine Hypokaliämie, z.B. infolge eines Aszites oder der Gabe bestimmter Diuretika. Niedrige Kaliumkonzentrationen werden durch einen Austausch von intrazellulärem Kalium gegen Natrium und H+-Ionen kompensiert. Dies führt zu einer extrazellulären Alkalose und einer intrazellulären Azidose. Infolgedessen kann das Ammoniak leicht die Zellmembran durchdringen, wird aber intrazellulär ionisiert, sodass es dann die Zelle nicht mehr verlassen kann und akkumuliert. Dies erklärt das Vorliegen niedriger Plasmakonzentrationen trotz ausgeprägter neurologischer Symptome.
Klinik
Die Klinik bei einer Hepatoenzephalopathie ist variabel und hängt von den metabolischen Störungen des Gehirns ab. Das Syndrom wird oft durch exo- oder endogene Faktoren wie z.B. reichliche Eiweißaufnahme oder Dehydratation ausgelöst.
Als charakteristisch gelten ein episodisch auftretendes abnormes Verhalten sowie zentralnervöse Störungen. Epileptische Anfälle kommen nur selten vor und treten häufig nur zusammen mit anderen Symptomen auf. Oft folgt nach einem oder mehreren symptomatischen Tagen eine Periode mit geringen oder ausbleibenden Symptomen. Diese Phase kann eine oder gar mehrere Wochen anhalten. Neben neurologischen Störungen sind auch nicht-neurologische Anzeichen wie z.B. Polyurie, Erbrechen, Diarrhö, Gewichtsverlust, Apathie und verminderte Belastbarkeit möglich.
| Neurologische Symptome | |
|---|---|
| Stadium 1: |
|
| Stadium 2: |
|
| Stadium 3: |
|
| Stadium 4: |
|
Diagnose
Die Diagnose wird durch eine erhöhte Ammoniakkonzentration im Plasma bestätigt.
In Zweifelsfällen ist ein Ammoniumchlorid-Toleranztest durchzuführen. Dieses Testverfahren kann man auch zur Diagnostik einer portosystemischen kollateralen Hepatoenzephalopathie verwenden. Auch bei Lebererkrankungen sollte das anfallende Ammoniak schnell in der Leber metabolisiert werden, sodass nur im Falle eines portosystemischen Shunts ein Anstieg im Plasma beobachtet werden kann.
Als pathognomonisch gilt auch der Nachweis von Ammoniumbiuratkristallen im Harnsediment (außer bei Dalmatinern und Englischen Bulldoggen).
Parallel dazu sind unterschiedliche bildgebende Verfahren (z.B. Röntgen, Ultraschall, Blutbild, CT/MRT u.ä.) indiziert.
Therapie
Die Therapie umfasst in erster Linie die Behandlung der auslösenden Grunderkrankung. Im Idealfall kann so eine vollständige Remission der Enzephalopathie erzielt werden.
Zusätzlich sind folgende Maßnahmen sinnvoll:
- Diät
- aromatische und verzweigtkettige Aminosäuren
- Lactulose
- Infusionstherapie
- symptomatische Maßnahmen
Um die Leber zu entlasten, ist eine proteinarme und kohlenhydratreiche Fütterung notwendig, z.B. z.B. mit kommerziellen Low-Protein-Futtermittel bzw. spezielle Leberdiäten. Aufgrund mangelnder Metabolisierung kommt es bei proteinreicher Ernährung zu einem Anstieg an aromatischen Aminosäuren (Tyrosin, Tryptophan und Phenylalanin) im Gehirn. Tyrosin ist ein Vorläufer von Katecholaminen wie Dopamin und Noradrenalin. Bei einem Tyrosinüberschuss bilden sich verstärkt Nebenprodukte (Tyramin, Octopamin), welche die Katecholaminrezeptoren blockieren. Durch die zusätzliche Gabe von verzweigtkettigen Aminosäuren (Leucin, Isoleucin und Valin) kann ein Teil dieser Rezeptoren freigehalten werden, sodass die Katecholamine binden und ihre physiologische Wirkung entfalten können.
Unterstützend sollte Lactulose (65%ige Lösung) in einer Dosierung von 1 bis 3 ml/kgKG (auf 3 mal täglich verteilt) oral verabreicht werden. Da Lactulose im Dünndarm nicht absorbiert wird, wird es im Kolon durch die Bakterienflora zu instabilen freien Fettsäuren abgebaut. Infolge dessen kommt es zu einer Übersäuerung, die zu einer Verschiebung in Richtung des nicht-absorbierbaren ionisierten Ammoniaks führt. Gleichzeitig wird die Darmmotilität erhöht und eine veränderte, weniger ammoniakbildende Flora gebildet. Falls durch die Diät und die Lactulosegabe keine genügende Besserung erzielt werden kann, können versuchsweise Antibiotika (z.B. Metronidazol 7,5 mg/kgKG, 2 mal täglich) zur Reduktion der ammoniakproduzierenden Bakterien verabreicht werden.
Bei akuten Symptomen ist eine intensive Infusionstherapie mit Mischinfusionen (physiologische NaCl-Lösung und 2,5%ige Glukoselösung mit Kalium-Zusatz) indiziert. Zusätzlich sollten Reinigungseinläufe mit warmem Wasser sowie Lactulose durchgeführt werden, um eine weitere Absorption intstinaler Enzephalotoxine zu verhindern. Ähnlich wie in der Humanmedizin kann versuchsweise auch der Benzodiazepinrezeptor-Antagonist Flumazenil (0,02 mg/kgKG i.v.) verabreicht werden. Bei Verdacht auf ein Hirnödem ist zusätzlich Mannitol (0,5 bis 1 mg/kgKG über 30 Minuten i.v.) indiziert.
Hinweis: Diese Dosierungsangaben können Fehler enthalten. Ausschlaggebend ist die Dosierungsempfehlung in der Herstellerinformation.
Literatur
- Kohn B, Schwarz G (Hrsg.). 2017. Praktikum der Hundeklinik. 12., aktualisierte Auflage. Stuttgart: Enke Verlag in Georg Thieme Verlag KG. ISBN: 978-3-13-219961-3