Morbus Gaucher









Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
Loslegennach dem französischen Arzt Philippe Charles Gaucher (1854-1918)
Synonym: Gaucher-Krankheit
Englisch: Gaucher's disease, Gaucher disease
Definition
Der Morbus Gaucher (sprich: [goˈʃe]) ist die häufigste lysosomale Speicherkrankheit aus dem Formenkreis der Sphingolipidosen, die durch eine genetisch bedingte Störung im Abbau der Lipidsubstanz Glukozerebrosid verursacht wird. Der Erkrankung liegt ein Mangel des lysosomalen Enzyms Glukozerebrosidase zugrunde.
Geschichte
1882 wurde die Krankheit erstmals von Philippe Charles Gaucher beschrieben, der in seiner Dissertation über eine Patientin mit Splenomegalie berichtete, 1905 wurde der Ausdruck Gaucher-Krankheit erstmals von N.E. Brill verwendet, der eine familiäre Häufung vermutete und einen autosomal-rezessiven Erbgang postulierte.
Klassifikation
Beim Morbus Gaucher werden verschiedene Verlaufsformen unterschieden:
Nicht-neuronopathische Verlaufsform (Typ 1)
Inzidenz: 1:40.000–60.000 / ca. 1:1000 bei Ashkenazi-Juden. Ausbruch in jedem Alter möglich.
Symptomatik beim Kind:
- Splenomegalie und Hypersplenismus, zusätzlich Anämie, Leuko- und Thrombopenie
- Skelettbeteiligung
- Auflockerungsherde an Phalangen, Kieferknochen und Wirbelkörpern (Schmerzen)
- „Erlenmeyer-Kolbenauftreibung“ am distalen Femur (80%)
- Hüftkopfnekrose (Jugendliche) durch vaskuläre Störungen
- Minderwuchs, Dystrophie
Siehe auch: Skelettbeteiligung bei Morbus Gaucher
Symptomatik beim Erwachsenen:
- Splenomegalie > 90% d.F.
- Pingueculae
- Hepatomegalie, selten mit Leberfunktionsstörung oder portaler Hypertonie
- Lungenbeteiligung mit diffusen, feinflächigen Infiltraten, Husten und rezidivierenden Pneumonien
- Lipidspeicherung in der Niere, meist symptomlos
- Bei älteren Patienten Disposition zu malignen Neoplasien (Morbus Hodgkin, Myelome)
Weitere Symptome, nicht altersspezifisch:
- Pulmonale Hypertonie unbekannter Genese, insbesondere nach erfolgter Splenektomie
- Anämie, Thrombozytopenie, Retikulozyten sind meist erhöht, Hämorrhagische Diathese
- Biochemische Veränderungen (bedingt durch gestörte Makrophagenaktivität): saure Phosphatase, ACE und Chitotriosidase erhöht
Akut neuronopathische Verlaufsform (Typ 2)
Inzidenz: < 1:100.000, panethnisch
Die Manifestation der akut-neuronopathischen Verlaufsform erfolgt im Säuglinsalter (2.-3. Lebensmonat).
Symptomatik:
- Fütterungsschwierigkeiten, Gedeihstörungen
- Gehäufte Atemwegsinfekte
- Hepatosplenomegalie → vorgewölbtes Abdomen
Ab dem 6. Lebensmonat:
- Neurologische Symptomatik
- Dysphagie, Stridor, Augenmuskellähmungen, selten Krampfanfälle
- Fortschreitender zerebraler Abbauprozess
- Finalstadium
- Ausgeprägte Kachexie
- Gelenkkontrakturen
- Therapieresistente Infektionen
Der Exitus erfolgt in der Regel zwischen dem 2. und 3. Lebensjahr. Vereinzelt treten neonatale Manifestationsformen auf, die nicht mit dem Leben vereinbar sind, beispielsweise die kongenitale Form mit Hydrops fetalis (wie bei anderen lysosomalen Speicherkrankheiten).
Chronisch neuronopathische Verlaufsform (Typ 3)
Inzidenz: < 1:50.000 bis 1:100.000, panethnisch, gehäuft bei Familien aus Nordschweden (Norbottrischer Typ)
Symptomatik:
Die chronisch neuronopathische Verlaufsform weist einen langsamen Verlauf auf, es handelt sich insgesamt um ein sehr heterogenes Krankheitsbild. Die Symptomatik beginnt meistens im 2. – 3. Lebensjahr (bei 30% auch später).
- Beginn häufig mit FUO
- Vermehrte Blutungsneigung (Hämorrhagische Diathese)
- Hepatosplenomegalie - abdominelle Beschwerden
- Panzytopenie (Knochenmarksinfiltration + Hypersplenismus)
- Störung der Augenmotilität (horizontale, supranukleäre Blickparese = Blickapraxie)
- Leichte mentale Retardierung, Verhaltensauffälligkeiten, Choreathethosen, Krampfanfälle
- Myoklonie als prognostisch ungünstiges Symptom
Ätiologie und Pathogenese
Der Morbus Gaucher ist eine hereditäre Sphingolipidose verursacht durch einen genetischen Defekt der β-Glukozerebrosidase, weltweit gibt es zwei Familien, die als seltene Ursache einen sogenannten Aktivatordefekt als Erkrankungsursache haben. (Saposin-C-Mangel).
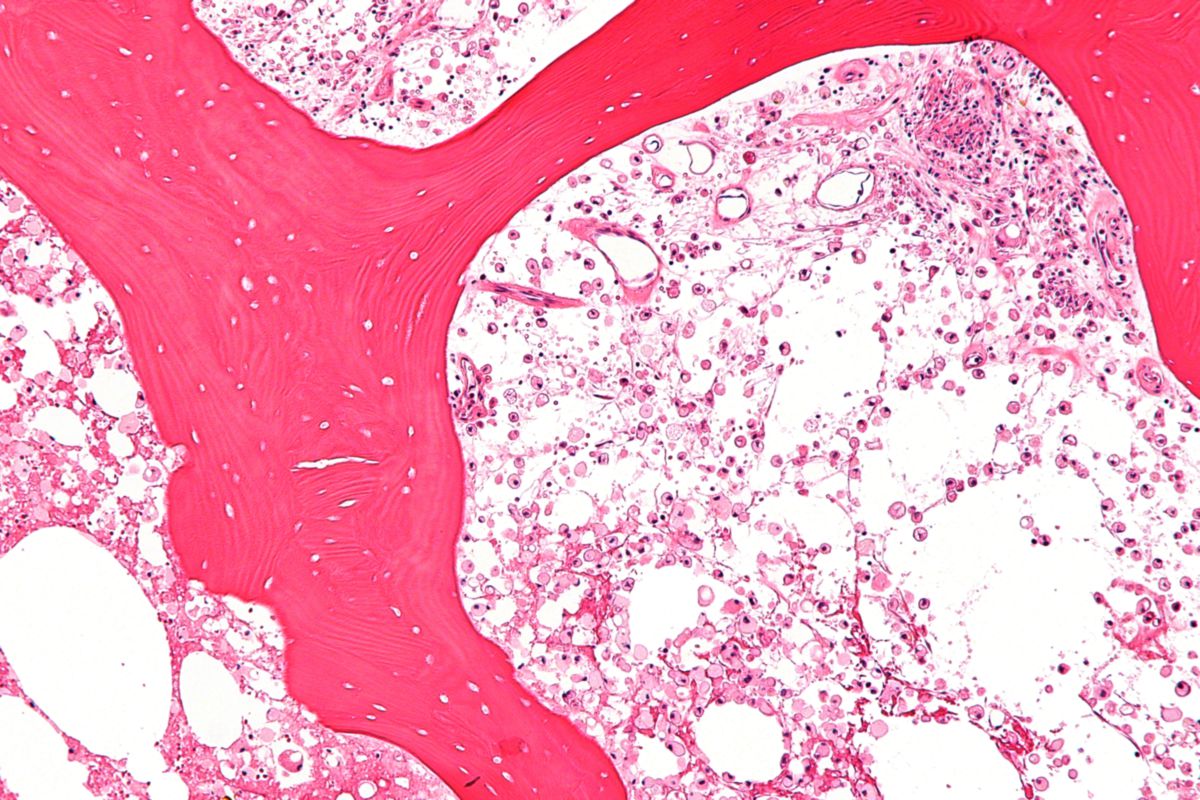
Die Glukozerebrosidase degradiert komplexe Glukosphingolipide, welche wesentliche Bestandteile der Zellmembranen sind. Glukozerebrosid (Substrat des Enzyms) kann nicht in Glukose und Ceramid gespalten werden. Es wird überwiegend in Makrophagen gespeichert (Entstehung von sogenannten „Gaucher-Zellen“), welche in Milz, Leber, Knochenmark, Lymphknoten und gelegentlich in der Lunge nachgewiesen werden können.
Der Glukozerebrosidgehalt in der Milz ist 10-1000-fach erhöht, was trotzdem nur < 2% des Gesamtorgangewichts ausmacht. Der Anstieg im Blutplasma ist weniger ausgeprägt.
Die Speicherung alleine ist nicht ausschlaggebend für die Symptomatik, es wird angenommen, dass es außerdem zu einer Aktivierung von Makrophagen mit Zytokin- und lysosomaler Proteinfreisetzung kommt (z.B. vermehrte Expression der Chitotriosidase, bis 4000x erhöht), welche von Gaucher-Zellen produziert werden. Die Plasmakonzentration dieser freigesetzten Substanzen korreliert mit der Zahl der Gaucher-Zellen im Körper.
Die neurologische Beteiligung ist bisher nur ungenügend geklärt worden. Im Vergleich zu anderen Lipidosen kommt es kaum zu ZNS-Ablagerungen, nur selten zu intraneuronalen Ablagerungen von Glukozerebrosid. Gaucher-Zellen können in den perivasculären Virchow-Robinsonschen Räumen und der tiefen weißen Substanz nachgewiesen werden.
Bei der akut-neuronopathischen Gaucher-Maus lagern sich Glukozerebroside diffus in Mikrogliazellen ab, vereinzelt finden sich Antisulfat-Antikörper, denen eine Bedeutung für die Neuropathie eingeräumt wird.
Neuronale Zelluntergänge finden sich in
Insgesamt fehlt jedoch eine klare Beziehung zwischen Einlagerungen und Neuronenuntergang.
Epidemiologie
Ein Morbus Gaucher kann in allen ethnischen Populationen gefunden werden, eine besonders hohe Prävalenz zeigt sich bei Ashkenazi-Juden für die nicht-neuronopathische Verlaufsform (1 : 400 - 1000). Die Prävalenz für Kaukasier beläuft sich auf ungefähr 1 : 50.000, wobei genaue Daten fehlen und viele Patienten unerkannt sein dürften.
Unterschiedliche Mutationen verursachen unterschiedliche Phänotyp-Ausprägungen.
Symptomatik
Die Symptomatik kann sehr heterogen ausgeprägt sein und ist zudem von Mutation zu Mutation unterschiedlich, so zeigt sich z.B. bei der bei Ashkenazi-Juden sehr häufigen Mutation N370S/N370S eine meist milde Ausprägung ohne Progression, manchmal werden sogar subjektiv symptomlose homozygote Familienmitglieder gefunden. Objektiv lassen sich bei allen homozygot Erkrankten Organmanifestationen (insbesondere Niere, Knochen) nachweisen.
Die homozygote Form der Mutation L444P, wie sie neben N370S auch häufig bei Europäern nicht-jüdischer Abstammung gefunden wird, führt hingegen oft zu einer neuronopathischen Verlaufsform mit rascher Progression und schweren viszeralen und neurologischen Komplikationen.
Leitsymptome
- Abgeschlagenheit
- Müdigkeit
- Knochenschmerzen
Häufige Symptome
- erhöhte Blutungsneigung
- Hypermenorrhoe
- Epistaxis
- Hautblutungen
- Nachblutungen bei Bagatelltraumen oder Operationen
- geburtshilfliche Komplikationen
- Metrorrhagien
- Zahnfleischbluten
- Oberbauchbeschwerden bei Hepatosplenomegalie
- erhöhte Infektneigung
Seltene Symptome
- häufige Zahnbehandlungen
- unklare Effloreszenzen
- okuläre Beschwerden (Fremdkörpergefühl, Visusminderung)
- Dyspnoe bei körperlicher Belastung (pulmonale Hypertonie)
Symptome bei Kindern
- Oberbauchbeschwerden
- Gedeihstörungen, Wachstumsretardierung
- Knochenschmerzen
Diagnostik
Anamnese
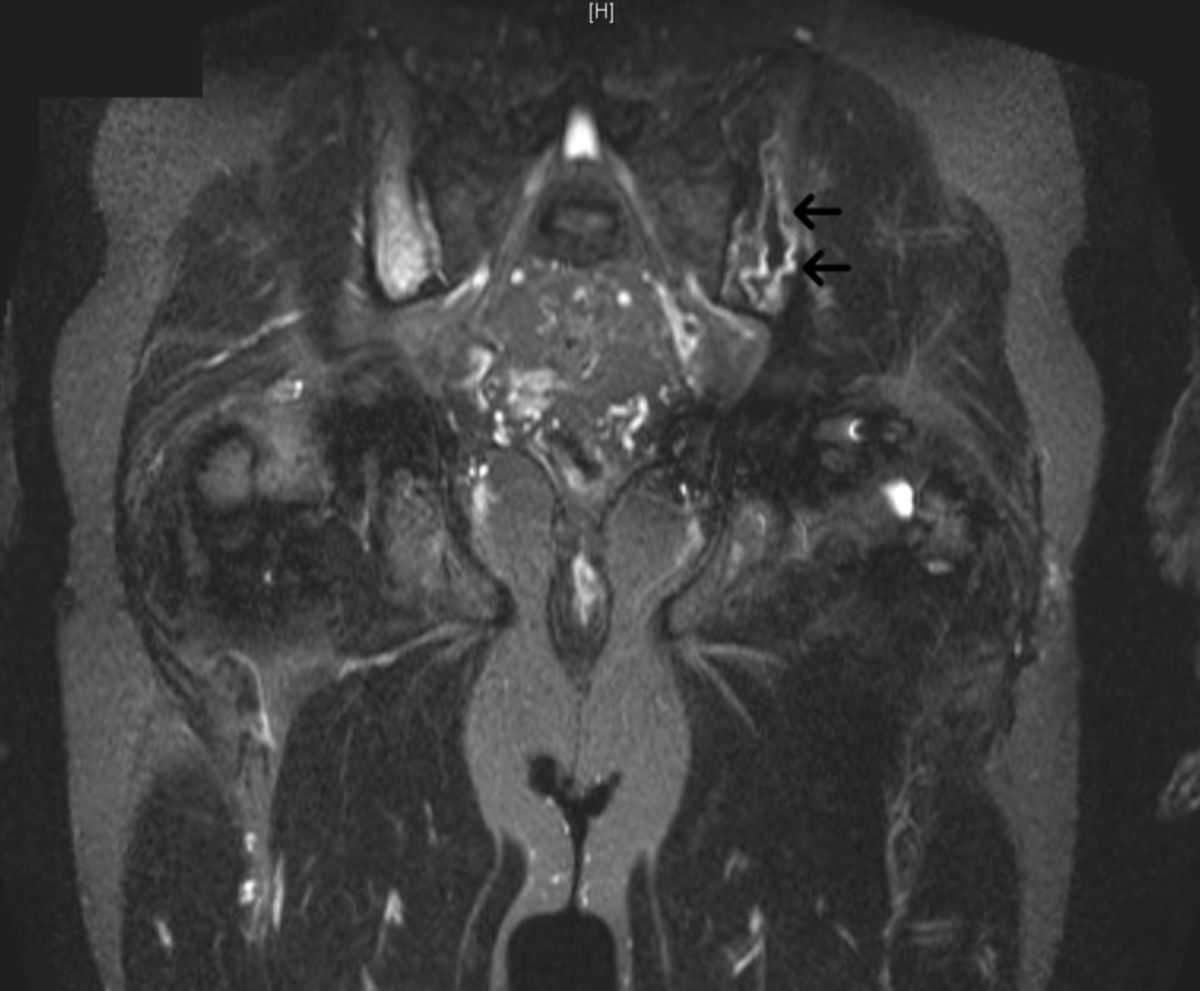
Die Anamnese sollte in erster Linie Fragen nach häufigen klinischen Befunden bei Morbus Gaucher beinhalten:
- Splenomegalie (ausgeprägt)
- Hepatomegalie (weniger ausgeprägt)
- pathologische Frakturen
- aseptische Knochennekrosen
- Osteolysen, Osteopenie, Osteoporose
- Milzinfarkte
- Anämie
- Thrombozytopenie
- Leukozytopenie
- Pseudotumoren
- Hautblutungen (Petechien, Sugillationen, Ekchymosen, Purpura)
Selten sind eine pulmonale Hypertonie, Leberzirrhose und Glaskörperinfiltrationen.
Ein Drittel der Patienten sind zum Zeitpunkt der ersten Vorstellung bereits splenektomiert.
Bei Kindern muss die mentale und statomotorische Entwicklung differenziert werden sowie neurologische bzw. neuroophtalmologische Auffälligkeiten (Strabismus, Krampfanfälle) erfragt werden.
Fragen zur Familienanamnese sollten umfassen: regionale Herkunft, Vorhandensein gleichartiger Symptome bei Familienmitgliedern, frühzeitige Todesfälle in der Familie. Eventuell kann die Erstellung eines Stammbaumes hilfreich sein.
Bei schweren Verlaufsformen empfiehlt sich eine zahnärztliche Untersuchung mit Erstellung eines Orthopantomogramms (Panoramaröntgen), da der Unterkieferknochen als langer Röhrenknochen häufig ähnliche Destruktionen wie Femur und Tibia aufweisen kann.
Labor
Anfangs oft milde Anämie und Thrombozytopenie <80.000/μl, später <20.000/ μl, bei ausgeprägter Hepatosplenomegalie findet sich eine Panzytopenie, häufig fehlt jedoch eine Leukozytopenie. Eine Blutungsneigung mit Petechien und Hämatomen ist schon bei geringer Thrombopenie möglich, da plasmatische Gerinnungsstörungen mit PTT-Verlängerung hinzukommen können.
Die Labordiagnostik erfüllt drei Funktionen:
- Ausschluss des Morbus Gaucher
- Sicherung der Diagnose
- Verlaufskontrolle
Neben einem Blutbild erfolgt auch die Messung nicht-tartrathemmbarer saurer Phosphatase, ACE, Lysozym (lysosomale Parameter) und des Ferritins für die Verlaufskontrolle.
Viele Patienten zeigen eine Erhöhung der Transaminasen und der Cholestaseparameter (AP, GGT), trotz Hepatomegalie (80%) sind Leberzirrhosen selten.
Eine Messung der Chitotriosidase sollte bei Verdacht immer erfolgen, da dieses Enzym bei Gaucher-Patienten 10x – 1000x erhöht sein kann und mit der Zahl von Gaucher-Zellen im Körper korreliert. Die Chitotriosidase dient zur Verlaufskontrolle, zur Therapieeinstellung und zur Erkennung von Rezidiven.
Liegt keine Erhöhung der Chitotriosidase bei einem unbehandelten Morbus Gaucher vor, so handelt es sich um einen Defekt des Chitotriosidasegens, was auf ca. 5% der Gaucher-Patienten zutrifft.
Lysosomale Verlaufsparameter sind in der Regel bei Gaucher-Patienten erhöht und daher von hohem prädiktivem Wert. Sind alle Parameter normal, ist ein Morbus Gaucher ausgeschlossen.
Bildgebung
Bildgebende Verfahren werden hauptsächlich zur Beurteilung der Skelettbeteiligung bei Morbus Gaucher hinzugezogen. Diese sind aus Übersichtsgründen in einem eigenen Kapitel beschrieben.
Diagnostisches Vorgehen
Aus EDTA- oder Heparinblut erfolgt die direkte Messung der β-Glukozerebrosidase in Leukozyten, bei Leukozytopenie auch aus kultivierten Fibroblasten (Hautbiopsie).
Bei typischem klinischem Bild und eindeutig erniedrigter Glukozerebrosidase-Aktivität ist die Diagnose gesichert. Ergänzend kann ein Nachweis des Gendefekts erbracht werden.
Der Nachweis von Gaucher-Speicherzellen in der Knochenmarksbiopsie ist kein Beweis für einen Morbus Gaucher, da diese auch bei Histiozytose, Thalassämie und granulomatösen Erkrankungen nachgewiesen werden können oder mit Schaumzellen beim Morbus Niemann-Pick verwechselt werden können.
Komplikationen
Die häufigsten Komplikationen ergeben sich aus Blutungen und Milzrupturen. In Japan verstarb ein Patient von 5 an Lungenversagen (bei Zustand nach Splenektomie, mit hepatischer und pulmonaler Beteiligung). Leberzirrhosen sind trotz der meist auftretenden Hepatomegalie eine seltene Komplikation.
Weiterhin wurde das gehäufte Auftreten von malignen Erkankungen, vor allem des lymphatischen Systems beobachtet (Plasmozytom, Lymphom etc.).
Lebenserwartung
Die chronisch-nichtneuronopathische Verlaufsform hat eine leicht bis mäßig erniedrigte Lebenserwartung, genaue Studien sind nicht vorhanden. Die Sterblichkeit wird durch Blutung, Milzruptur und Malignome bestimmt.
Die akut neuronopathische Form verläuft innerhalb von 1-2 Jahren nach Geburt letal.
Zur Lebenserwartung bei der chronisch-neuronopathischer Form gibt es keine großen Studien. ZNS-Komplikationen und schwere viszerale Komplikationen prägen das Krankheitsbild, die Lebensqualität ist durch die Knochenbeteiligung mit Frakturen und Femurkopfnekrosen stark herabgesetzt und fesselt die Patienten oft an den Rollstuhl.
Therapie
Enzymersatztherapie (ERT)
Die i.v.-Gabe des nicht modifizierten Placentaenzyms führte nicht zu einer wesentlichen Besserung, obwohl die Glukozerebrosid-Konzentrationen in Leber und Blut gesenkt werden.
Bei der weiteren Forschung stellte sich heraus, dass das nicht-modifizierte Enzym sich gleichmäßig im Organismus verteilt und relativ wenig in Makrophagen gelangt und daher dort kaum zur Wirkung kommt.
Nach weiteren neuen Erkenntnissen darüber, dass Makrophagen Glykoproteine durch einen Rezeptor aufnehmen, der relativ spezifisch für Mannose und β-N-Azetylglukosaminidase ist, gelang es, das Enzym biochemisch so zu modifizieren, dass außer der Mannosekette alle anderen Zuckerseitenketten des Enzyms abgespalten wurden (Alglucerase), was zu einer nahezu „selektiven“ Aufnahme der Glukozerebrosidase in Makrophagen führt.
Seit 1991 ist die Therapie mit Alglucerase zugelassen, verwendet wird derzeit die rekombinant hergestellte Imiglucerase (Cerezyme®). Sie kann in genügend hoher Menge hergestellt werden und birgt nicht das theoretische Restrisiko einer Infektion wie bei der Alglucerase. Im Allgemeinen ist die Enzymersatztherapie sowohl gut wirksam als auch verträglich, jedoch auch mit hohen Kosten verbunden.
Substratreduktion
Diese Therapieform zielt darauf ab, die Synthese der Speichersubstanz durch Enzyminhibitoren partiell zu hemmen. Derzeit (2026) sind die Arzneistoffe Eliglustat und Miglustat in Deutschland zugelassen.
Begleittherapien
Die Begleittherapie umfasst die Behandlung und Begleitung von krankheitsbedingten Komplikationen. Sie umfasst unter anderem:
- Prothethischer Gelenkersatz nach ausreichend langer Enzymersatztherapie
- sonstige orthopädische Maßnahmen
- Osteoklastenhemmstoffe (Bisphosphonate)
- Antiepileptische Therapie
- Sorgfältige Krebsvorsorgeuntersuchungen
- Bei splenektomierten Patienten
- Impfung gegen Haemophilus influenzae und gegen Pneumokokken
- Notfallausweis sowie Stand-by-Antibiotikum auf Reisen
- Physikalische Therapie sowie leichte, regelmäßige körperliche Betätigung
- Eine Splenektomie ist bei früher Anbehandlung mit der Enzymersatztherapie in der Regel nicht erforderlich