Sichelzellanämie








Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenSynonyme: Sichelzellenanämie, homozygote Sichelzellerkrankung, Drepanozytose
Englisch: sickle cell anaemia
Definition
Die Sichelzellanämie ist eine Erbkrankheit, die zu den hämolytischen Anämien bzw. Hämoglobinopathien gehört. Sie wird durch einen genetischen Defekt (Punktmutation) ausgelöst, der zur Bildung von irregulärem Hämoglobin, dem sogenannten Sichelzellhämoglobin (Hämoglobin S, HbS) führt.
Genetik
Die Sichelzellanämie folgt einem autosomal-rezessiven Erbgang. Es besteht eine Punktmutation der ß-Globinkette, wobei an Position 6 hydrophiles Glutamat gegen hydrophobes Valin substituiert wurde. Dadurch werden hydrophobe Wechselwirkungen zwischen den ß-Globinketten ermöglicht. Im desoxygenierten Zustand polymerisiert HbS zu langen Faserstrukturen, die zur Hämolyse führen. Diese Polymerisation stellt den molekularen Schlüsselschritt der Erkrankung dar. Sie führt zu einer verminderten Verformbarkeit, zu Membranschäden und zur Dehydratation der Erythrozyten. In der Folge kommt es zu Hämolyse und Vasookklusion.
Die Sichelzellanämie im engeren Sinn betrifft homozygote Merkmalsträger mit HbSS, bei denen sowohl das mütterliche als auch das väterliche Allel verändert ist. Heterozygote Träger besitzen dagegen nur ein HbS-Allel und weisen das Sichelzellmerkmal auf. Sie sind in der Regel klinisch unauffällig. Aufgrund der Kodominanz beider Allele wird bei heterozygoten Merkmalsträgern sowohl normales als auch verändertes Hämoglobin gebildet. Allerdings wird auch bei ihnen unter starkem Sauerstoffmangel, wie zum Beispiel während einer Narkose, die Sichelform der Erythrozyten ausgebildet, sodass es zu einer Beeinträchtigung der Organdurchblutung kommen kann. Compound-Heterozygote, das heißt Patienten mit einem HbS-Allel und einem weiteren pathologischen Allel (z.B. HbC, HbE), zeigen ein der Sichelzellanämie ähnelndes Krankheitsbild (Sichelzellsyndrom).
Epidemiologie
Die Sichelzellanämie hat die größte Verbreitung in den Malariagebieten Afrikas, des Mittelmeerraums, des Nahen Ostens und Indiens. In Äquatorialafrika sind 25 bis 40 % der Bevölkerung heterozygote Merkmalsträger. Die Häufigkeit des Defekts nimmt mit dem Abstand zum Äquator deutlich ab. Bei der afroamerikanischen Bevölkerung Amerikas liegt die Häufigkeit nur noch zwischen 5 und 10 %.
Dieses Phänomen lässt sich dadurch erklären, dass heterozygote Merkmalsträger eine relative Resistenz gegen Malaria besitzen – ein Umstand, der in Malariagebieten einen deutlichen Selektionsvorteil darstellt. In gemäßigten Breiten ist der Selektionsvorteil aufgrund der fehlenden Malaria nicht wirksam.
Weltweit werden jährlich mehr als 400.000 Kinder mit Sichelzellanämie geboren. Die Geburtsprävalenz ist regional unterschiedlich und in Gebieten mit hoher Malariaendemie am höchsten.
In Deutschland ist die Sichelzellkrankheit insgesamt selten, gewinnt jedoch zunehmend an Bedeutung in der medizinischen Versorgung. Betroffen sind vor allem Menschen mit familiärer Herkunft aus Endemiegebieten. Durch das seit 2021 etablierte Neugeborenenscreening werden betroffene Kinder frühzeitig diagnostiziert.
Klinik
Die Sichelzellanämie manifestiert sich in der Regel erst ab dem 6. Lebensmonat nach Abfall des fetalen Hämoglobins (HbF), das zuvor in den Erythrozyten vorherrscht. Die klinische Ausprägung ist interindividuell sehr variabel. Die Erkrankung ist eine chronische Multiorganerkrankung, die mit akuten vasookklusiven Komplikationen sowie progredienten Endorganschäden verbunden ist.
Das akute Krankheitsgeschehen der Sichelzellanämie findet während einer sogenannten Sichelzellkrise statt, die mit akuten Schmerzen und Angstzuständen verbunden ist. Die Häufigkeit und Schwere der Sichelzellkrisen variieren stark. Die Schmerzen können überall auftreten und wenige Stunden bis zwei Wochen anhalten. Eine frühe Manifestation im Kindesalter ist die Daktylitis im Sinne eines Hand-Fuß-Syndroms.
Auslöser
Mögliche Auslöser von Sichelzellkrisen sind:
- Infektionen, Fieber
- Hypoxie (beispielsweise auch beim Fliegen in großer Höhe)
- Dehydratation
- Azidose
- Hämolyse durch Medikamente, Kontrastmittel
- große körperliche Anstrengung
- Kälte
- Stress
Organschäden
Vasookklusionen bei Sichelzellanämien führen zu wiederholten Mikroinfarzierungen, die wiederum Organschäden nach sich ziehen. Bereits in den ersten Lebensjahren kommt es zur funktionellen Asplenie mit erhöhter Infektneigung. Dadurch besteht insbesondere ein erhöhtes Risiko für schwere bakterielle Infektionen durch bekapselte Erreger. Die akute venöse Obstruktion der Milz (splenische Sequestrationskrise) ist eine lebensbedrohliche Komplikation. Eine weitere akute hämatologische Komplikation ist die aplastische Krise, meist im Zusammenhang mit einer Parvovirus-B19-Infektion.
Die Obstruktion der Netzhautgefäße führt zu einer Sichelzellretinopathie. Nierenpapillennekrosen bedingen eine Hyposthenurie bzw. Isosthenurie, Proteinurie bis hin zur chronischen Nierenerkrankung und zum Nierenversagen. Ischämien von Knochen und Gelenken resultieren in aseptischen Nekrosen, chronischer Arthropathie und Anfälligkeit für Osteomyelitiden.
Das Hand-Fuß-Syndrom entsteht durch schmerzhafte Infarkte in den Fingern. Eine Infarzierung der venösen Ausstrombahn im Penis kann zum Priapismus mit bleibender Impotenz führen. Chronische Ulzerationen der Unterschenkel beruhen auf Ischämien der Haut, meist mit zusätzlicher Superinfektion. Ausdruck einer Sichelzellkrise der Lunge ist das akute Thoraxsyndrom mit Schmerzen, Tachypnoe, Fieber, Husten und Hypoxämie. Dabei begünstigt die verminderte arterielle Sauerstoffsättigung die Progression bis hin zur pulmonalen Hypertonie und zum Cor pulmonale.
Schlaganfälle treten bereits bei Kindern auf. Zusätzlich kommen stille zerebrale Infarkte vor, die mit neurokognitiven Einschränkungen assoziiert sein können. Chronische subakute Schäden des ZNS zeigen sich oft durch subtile Verhaltensänderungen und Leistungsabfall.
Weiterhin führt die hämolytische Anämie neben den typischen Anämiesymptomen zu multiplen Komplikationen (z.B. Ikterus, Gallensteine).
Labordiagnostik
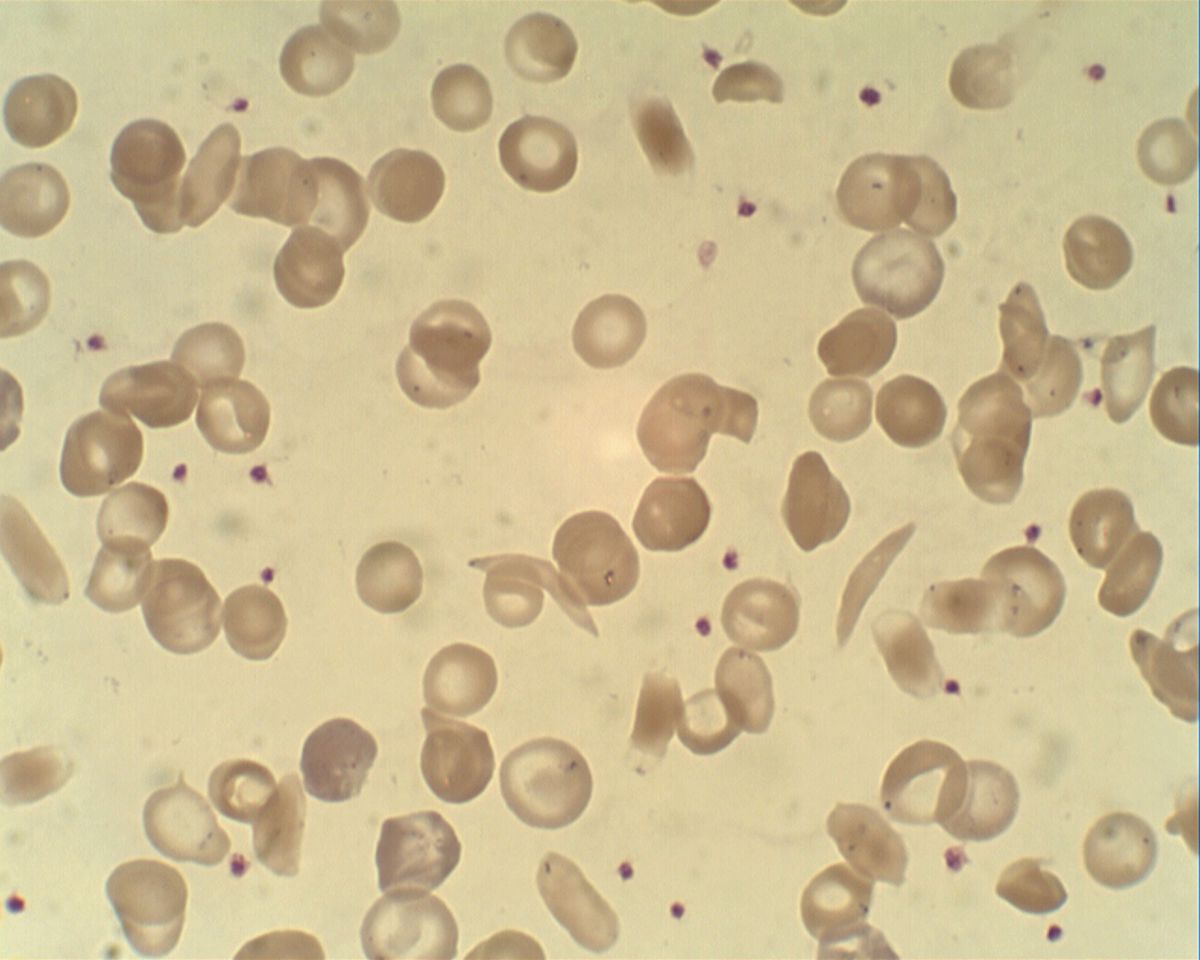
Infolge der hämolytischen Anämie sind Hämoglobin-Konzentration und Hämatokrit deutlich erniedrigt. Begleitend findet sich meist eine Retikulozytose und eine Granulozytose. Typische Laborzeichen der chronischen Hämolyse sind außerdem ein erhöhtes indirektes Bilirubin, eine erhöhte LDH und ein erniedrigtes Haptoglobin. Im Blutausstrich sind Sichelzellen erkennbar, gelegentlich auch Howell-Jolly-Körperchen als Zeichen einer gestörten Milzfunktion.
Der definitive Nachweis einer Sichelzellanämie erfolgt durch Hämoglobinelektrophorese, Massenspektrometrie und Sichelzelltest. Der Sichelzelltest ist als alleinige Diagnostik nicht ausreichend und im Neugeborenenalter wegen des hohen Anteils an fetalem Hämoglobin nur eingeschränkt verwertbar. Auch nach Bluttransfusionen kann die Interpretation der Hämoglobinfraktionen erschwert sein.
Die molekulargenetische Untersuchung kann die Diagnose bestätigen und ist insbesondere bei unklaren Befunden, zur Genotypisierung oder im Rahmen der Familienberatung sinnvoll. Auch eine Präimplantationsdiagnostik ist möglich.
Das Neugeborenenscreening auf Sichelzellanämie ist eine wichtige Untersuchung, die seit 2021 in Deutschland Teil der erweiterten Neugeborenen-Früherkennung ist. Durch eine Fersenblutprobe, die zwischen dem 3. und 5. Lebenstag entnommen wird, können betroffene Kinder frühzeitig erkannt werden. Dies ermöglicht eine rasche Einleitung prophylaktischer und unterstützender Maßnahmen und kann lebensbedrohliche Komplikationen verhindern.
Therapie
Die therapeutischen Maßnahmen richten sich nach Schweregrad, Genotyp, Organbeteiligung und Komplikationen der Erkrankung. Die Behandlung umfasst Basismaßnahmen, krankheitsmodifizierende medikamentöse Therapie, Transfusionsverfahren sowie kurative Therapieansätze.
Standardtherapie
Zu den Basismaßnahmen gehören eine ausreichende Hydratation, die Vermeidung von Hypoxie, eine konsequente Infektionsprophylaxe einschließlich Impfungen sowie die frühzeitige Behandlung von Infektionen.
Die symptomatische Therapie umfasst:
- Flüssigkeitssubstitution
- Analgetika (z.B. Paracetamol, Ibuprofen, Opioide)
- Antibiotika bei entsprechender Indikation.
Die wichtigste krankheitsmodifizierende medikamentöse Therapie ist Hydroxycarbamid. Es steigert die Bildung von fetalen Hämoglobins (HbF) und senkt die Frequenz von Schmerzkrisen und akutem Thoraxsyndrom. In aktuellen Leitlinien (2026) wird es bei vielen Patienten mit HbSS und verwandten schweren Genotypen als Standardtherapie empfohlen. Transfusionstherapeutische Maßnahmen sind:
- Erythrozytentransfusion
- Austauschtransfusion (Partielle Blutaustauschtransfusion, Erythrozytapherese)
- chronisches Transfusionsprogramm
Dabei ist zwischen einfacher Transfusion und Austauschtransfusion zu unterscheiden. Eine einfache Transfusion verbessert die Sauerstofftransportkapazität, ist jedoch durch das Risiko einer Hyperviskosität limitiert. Ein Hämoglobinwert von etwa 10 g/dl sollte in der Regel nicht überschritten werden.
Wichtige Indikationen für eine Austauschtransfusion sind unter anderem Schlaganfall, schweres akutes Thoraxsyndrom, Multiorganversagen sowie bestimmte chronische Indikationen wie die primäre oder sekundäre Schlaganfallprävention. Schmerzkrisen allein sind in der Regel keine Indikation zur Transfusion.
Chirurgische Maßnahmen umfassen in Einzelfällen:
- Splenektomie
- Cholezystektomie (bei Gallensteinen)
Ein kurativer Therapieansatz ist die allogene Stammzelltransplantation. Sie ist bislang die etablierteste kurative Standardoption und sollte insbesondere bei geeigneten Patienten mit HLA-kompatiblem Spender frühzeitig in einem erfahrenen Transplantationszentrum geprüft werden.
Zu den nicht mehr aktuellen medikamentösen Therapieoptionen zählen:
- Crizanlizumab ist in der Europäischen Union seit 2023 nicht mehr zugelassen.
- Voxelotor ist in der Europäischen Union weiterhin suspendiert und derzeit keine reguläre Therapieoption.
In den USA ist Crizanlizumab, ein gegen P-Selektin gerichteter monoklonaler Antikörper, weiterhin (2026) zur Reduktion der Häufigkeit vasookklusiver Krisen bei Sichelzellkrankheit zugelassen. In der EU wurde die Zulassung 2023 widerrufen, nachdem die Phase-III-Studie STAND keinen klinischen Nutzen gegenüber Placebo bestätigen konnte. [1]
Gentherapie
Einen neuen Therapieansatz bietet das Gentherapeutikum Exagamglogen-Autotemcel. Bei diesem Verfahren werden autologe hämatopoetische Stammzellen des Patienten mithilfe des CRISPR/Cas-Systems genetisch so modifiziert, dass sie vermehrt fetales Hämoglobin (HbF) produzieren. Dadurch lässt sich die Symptomatik deutlich verbessern.
Exagamglogen-Autotemcel ist in der Europäischen Union zur Behandlung von Patienten ab 12 Jahren mit schwerer Sichelzellkrankheit und rezidivierenden vasookklusiven Krisen zugelassen, sofern sie für eine Stammzelltransplantation geeignet sind und kein HLA-kompatibler verwandter Spender verfügbar ist. Der Arzneistoff wurde im Dezember 2023 vom CHMP der Europäischen Arzneimittelagentur (EMA) zur Zulassung empfohlen. Eine Zulassung durch die FDA erfolgte im gleichen Monat.[2]
In den USA ist mit Lovotibeglogen-Autotemcel eine weitere Gentherapie zur Behandlung der Sichelzellkrankheit zugelassen. [3] Diese ist derzeit (2026) jedoch nicht in der Europäischen Union verfügbar.
Verlaufskontrolle
Wichtige Maßnahmen sind die transkranielle Dopplersonographie zur Schlaganfallprävention und ein regelmäßiges Screening auf Organschäden.
Literatur
- Manger B et al.: Checkliste Rheumatologie. 4. Auflage, 2012. Thieme Verlag
- Schifferli J et al.: Internistische Notfälle: Sicher durch die Akutsituation und die nachfolgenden 48 Stunden. 8. Auflage, 2008. Thieme Verlag
- Ploier R: Differenzialdiagnosen in der Kinder- und Jugendmedizin. 1. Auflage, 2012. Thieme Verlag
- Onkopedia Leitlinie Sichelzellkrankheiten, abgerufen am 17. April 2026
- GeneReviews: Sickle Cell Disease, abgerufen am 17. April 2026
- Global burden of sickle cell disease, Lancet Haematology, 2023
- G-BA: Neugeborenenscreening auf Sichelzellkrankheit, abgerufen am 17. April 2026
- EMA: Widerruf der Zulassung von Crizanlizumab (Adakveo), abgerufen am 17. April 2026
- EMA: Voxelotor (Oxbryta) – Referral procedure, abgerufen am 17. April 2026
- FDA: Approval of gene therapies for sickle cell disease (Casgevy & Lyfgenia), abgerufen am 17. April 2026
- Gene therapy in sickle cell disease, 2024
Quellen
- ↑ EMA: Revocation of authorisation of sickle cell disease medicine Adakveo, abgerufen am 17.04.2026
- ↑ Full Prescribing Information Casgevy, FDA. Abgerufen am 19.12.2023
- ↑ LYFGENIA, abgerufen am 17.04.2026