Alzheimer-Krankheit










Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
Loslegennach dem deutschen Neurologen Alois Alzheimer (1864 bis 1915)
Synonyme: präsenile Demenz (veraltet), Demenz vom Alzheimer-Typ, Alzheimer-Demenz, Morbus Alzheimer
Englisch: alzheimer's disease
Definition
Die Alzheimer-Krankheit ist eine häufige neurodegenerative Erkrankung, die durch eine progrediente Atrophie der Großhirnrinde (Cortex cerebri) mit charakteristischen neuropathologischen und neurochemischen Veränderungen gekennzeichnet ist. Die Betroffenen entwickeln eine über Jahre zunehmende Demenz, die in späteren Krankheitsstadien zu einem Verlust der Alltagskompetenz und zu einem Persönlichkeitszerfall führt.
ICD10-Codes
- F00.-* Demenz bei Alzheimer-Krankheit (G30.-+)
- F00.0* Demenz bei Alzheimer-Krankheit, mit frühem Beginn (Typ 2) (G30.0+)
- F00.1* Demenz bei Alzheimer-Krankheit, mit spätem Beginn (Typ 1) (G30.1+)
- F00.2* Demenz bei Alzheimer-Krankheit, atypische oder gemischte Form (G30.8+)
- F00.9* Demenz bei Alzheimer-Krankheit, nicht näher bezeichnet (G30.9+)
Epidemiologie
Frauen sind von der Alzheimer-Krankheit häufiger betroffen als Männer. Die durchschnittliche Krankheitsdauer beträgt etwa acht Jahre bis zum Tod. Die Prävalenz steigt mit zunehmendem Alter an und liegt bei etwa 1 % in der Gesamtbevölkerung, bei etwa 5 bis 10 % bei den über 65-Jährigen und bei etwa 30 % bei den über 80-Jährigen.[1]
Einteilung
- Typ 1: Alzheimer-Krankheit mit spätem Beginn, d.h. nach 65 Jahren
- Typ 2: Alzheimer-Krankheit mit frühem Beginn, d.h. unter 65 Jahren (selten, <10 %)
Ätiologie
Die genaue Ätiologie der Alzheimer-Krankheit ist bislang (2026) noch nicht geklärt. Es handelt sich um eine degenerative Erkrankung mit einer diffusen Atrophie der Hirnrinde (im späteren Verlauf auch des Marklagers). Betroffen sind hier insbesondere der Temporallappen, der Parietallappen und der Hippocampus. Die Gehirnschrumpfung kann bis zu 20 % betragen.
Histopathologische Untersuchungen zeigen, dass Ablagerungen des Proteins Beta-Amyloid (Aβ) im Hirnparenchym und in den Hirngefäßen ein wesentlicher Treiber der Erkrankung sind. Die Erkrankungsmechanismen haben dabei Ähnlichkeit mit denen der Creutzfeldt-Jakob-Erkrankung (CJD), die durch Prionen ausgelöst wird. Wie die CJD wurde auch die Alzheimer-Krankheit in der Vergangenheit in einigen Fällen iatrogen durch Verabreichung von kontaminierten humanen Wachstumshormon-Präparaten übertragen. In diesen Präparaten ließ sich nachträglich Beta-Amyloid nachweisen.[2]
Genetik
Genetische Faktoren spielen eine wichtige Rolle bei der Entstehung der Alzheimer-Krankheit. Der ApoE-ε4-Genotyp ist einer der wichtigsten genetischen Risikofaktoren für die Enstehung einer Alzheimer-Krankheit. Das sehr seltene Allel ApoE-ε2 wirkt hingegend protektiv.[3][4]
Unter 3 % aller Fälle von Alzheimer-Krankheit werden autosomal-dominant vererbt. Diese Form bezeichnet man als familäre Alzheimer-Krankheit. Bekannt sind ursächliche Mutationen im APP-Gen, Präsenilin-1-Gen und Präsenilin-2-Gen.[5][6][7]
Histopathologie
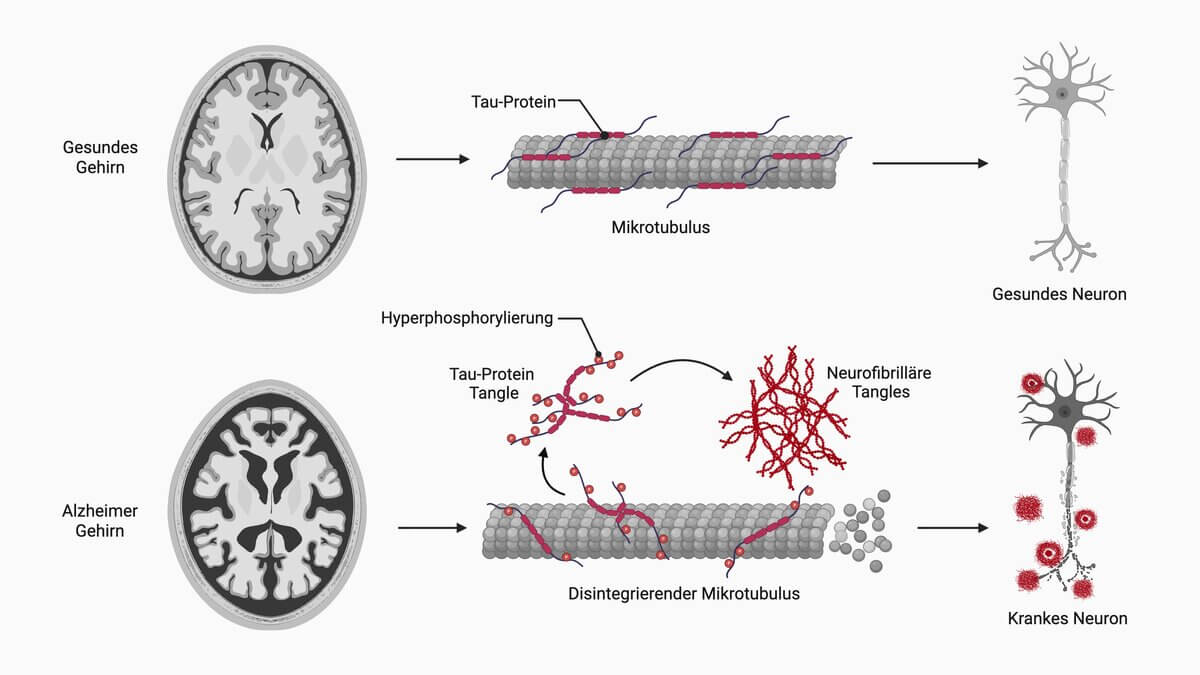
Mikroskopisch zeigen sich intrazelluläre Aggregate des Tau-Proteins (Neurofibrilläre Tangles, Alzheimer-Fibrillen), das ein wichtiges Strukturprotein zur Stabilisierung der Mikrotubuli in Neuronen ist. Dieser Aggregation liegt eine übermäßige Phosphorylierung der Tau-Proteine zugrunde. Ungeklärt ist, ob diese Phosphorylierung sekundärer Natur oder krankheitsauslösend ist. Gehen die betroffenen Neurone zu Grunde, erscheinen die neurofibrillären Tangles extrazellulär als sogenannte Ghost-Tangles.
Des Weiteren sind Ablagerungen extrazellulärer Beta-Amyloid-Plaques (senile Plaques) und eine zerebrale Amyloidangiopathie (Kongorot-Färbung) nachweisbar. Diese beiden mikroskopischen Korrelate der Alzheimer-Krankheit basieren auf der Ablagerung von Beta-Amyloid.[8]
Eine histopathologische Klassifikation auf Basis des Verteilungsmusters von Tau-Proteinen und Beta-Amyloid-Plaques bieten die Braak-Stadien.
Innerhalb der Neurone sieht man granuläre und vakuoläre Degenerationen, die Transmitterausschüttung von Acetylcholin ist durch Degeneration des Nucleus basalis vermindert. Diese Veränderungen sind allerdings nicht spezifisch und lassen sich auch in den Gehirnen nicht-dementer Personen nachweisen.
Darüber hinaus wird eine Verbindung zwischen dem Protein IGFBP7 und der Alzheimer-Krankheit vermutet. Im Mausmodell gingen erhöhte Konzentrationen von IGFBP7 mit einer verminderten Gedächtnisleistung einher.[9]

Symptomatik
Die Alzheimer-Krankheit geht mit einem Abbau des Gedächtnisses sowie kognitiver, emotionaler und sozialer Fähigkeiten einher. Das erste Symptom der Alzheimer-Krankheit ist eine zunehmende Störung der Merkfähigkeit und der Orientierung. Im weiteren Verlauf treten Perseverationen sowie Apraxie und Aphasie auf. Die Persönlichkeitsstruktur bleibt auch mit fortschreitender Erkrankung über einen langen Zeitraum erhalten.
| Fachausdruck | Klinik |
|---|---|
| Amnesie | Gedächtnisverlust, Vergesslichkeit |
| Apraxie | Verlust praktischer Fähigkeiten, z.B. Ankleiden, Schuhe binden, Kaffee kochen |
| Sensorische Aphasie | Fehlendes Sprachverständnis |
| Agnosie | Mangelnde Interpretationsfähigkeit wahrgenommener Informationen, Überforderung beim Treffen von Entscheidungen |
| Motorische Aphasie | Sprechstörung, unverständliche Sprache |
| Apathie | Mangelnde Anteilnahme |
Phänotypen
Vereinfacht unterscheidet man zwischen folgenden Phänotypen:
- amnestische Variante (häufigste Form): fortschreitendes amnestisches Syndrom vom "Hippocampus-Typ"
- posteriore kortikale Atrophie (PCA, Benson-Syndrom): Fortschreitende Störung der visuellen und ggf. anderer kognitiver Funktionen. Typische klinische Zeichen sind eine visuelle Agnosie und Apraxie.
- logopenische Variante der primär progressiven Aphasie (lvPPA): fortschreitende Beeinträchtigung beim Abruf von Einzelwörtern und der Wiederholung von Sätzen
- behavioral-dysexekutive Variante: fortschreitende Apathie oder Verhaltensenthemmung und stereotypes Verhalten oder fortschreitende vorherrschende exekutive Dysfunktionen
- kortikobasales Syndrom: fortschreitende asymmetrische Gliedersteifigkeit oder -akinesie, Dystonie, Myoklonie, orobukkale oder Gliedmaßenapraxie, kortikales sensorisches Defizit, Alien-Limb-Phänomen
- andere Varianten der primär progressiven Aphasie (PPA): semantische oder nicht flüssige PPA-Varianten
Verlauf
Der Verlauf der Alzheimer-Krankheit ist chronisch progredient und lässt sich grob in drei Stadien einteilen:
- Stadium 1: Amnesie
- Stadium 2: Apraxie, sensorische Aphasie, Agnosie
- Stadium 3: Bettlägerigkeit, Apathie, Inappetenz, Inkontinenz, motorische Aphasie
Jedes Stadium dauert ca. drei Jahre lang an.
Diagnostik
Überblick
- Eigenanamnese
- Fremdanamnese (Befragung der Angehörigen)
- Neuropsychologische Testverfahren (z.B. DemTect, Mini-Mental-Status-Test (MMST), Uhrentest)
- Liquordiagnostik: erhöhtes Tau-Protein und phosphoryliertes Tau-Protein, erniedrigtes β-Amyloidprotein Aβ (1-42) und erniedrigter Quotient Aβ (1-42)/Aβ (1-40)
- Molekularbiologisch (ApoE4-Genotyp)
- Radiologie: Hirnatrophie (betont temporal und hippocampal), dient dem Ausschluss von Differenzialdiagnosen
- Computertomografie (CT)
- Magnetresonanztomographie (MRT)
- Nuklearmedizin: Ein Diagnoseverfahren zur Früherkennung des Morbus Alzheimer ist die Software-gestützte Messung der Stoffwechselaktivität im Hippocampus mittels Positronenemissionstomographie (PET).
Radiologie
Bei der Alzheimer-Erkrankung finden sich MR-tomographisch die ersten atrophischen Veränderungen entorhinal (Brodmann-Areal 28), gefolgt vom Hippocampus, den Amygdala und dem Parahippocampus. Andere Strukturen innerhalb des limbischen Systems (z.B. der posteriore cinguläre Kortex) sind ebenfalls bereits früh betroffen. Anschließend kommt der temporale Neokortex hinzu sowie später zusätzlich symmetrisch alle neokortikalen Assoziationsbahnen.
Bildmorphologisch unterscheidet man vier Varianten der Alzheimer Erkrankung:[10]
- klassische Variante mit dem Atrophiemuster Hippocampus > temporal > parietal > frontal
- limbisch-prädominante Variante: Atrophie nur Hippocampus und entorhinal
- Hippocampus-aussparende Variante: entspricht der posterioren kortikalen Atrophie. Sie betrifft vorwiegend den Precuneus und den posterioren Gyrus cinguli.
- MR-bildgebend visuell weitgehend unauffällige Variante mit minimaler Atrophie
Bereits bei klinisch kaum auffälligen Alzheimer-Patienten ist das entorhinale Volumen bei den ersten beiden Varianten um 20 bis 30 % reduziert, wohingegen der Hippocampus in diesem Stadium nur um 15 - 25 % reduziert ist.[11] Hilfreich im Frühstadium ist insbesondere der ERICA-Score. Bei fortgeschrittener Demenz können jedoch auch Patienten mit einer FTD einen pathologischen Wert im ERICA-Score erreichen. Der MTA-Score ist weniger sensitiv als der ERICA-Score, da kein direkter Bezug zu dem beim Morbus Alzheimer als erstes betroffenen Hirnbereich (entorhinaler Kortex) besteht. Der Koedam-Score ist das Mittel der Wahl zur Beurteilung der biparietalen Atrophie bei der Hippocampus-aussparenden Variante.
Nuklearmedizin
Nuklearmedizinische Verfahren ermöglichen die Erfassung funktioneller Veränderungen und spezifischer molekularer Zielstrukturen bei neurodegenerativen Erkrankungen. Bereits viele Jahre vor dem Auftreten erster kognitiver Symptome können Amyloidablagerungen dargestellt werden. Reduktionen im Glukosestoffwechsel sowie pathologische Tau-Ablagerungen treten erst in späteren Krankheitsstadien auf.
Die FDG-PET ist ein etabliertes Diagnosewerkzeug für neurodegenerative Erkrankungen. Reduktionen im Glukosestoffwechsels lassen sich oft schon vor morphologischen Veränderungen nachweisen. Deutliche Stoffwechselveränderungen, die eine differenzierte Diagnosestellung ermöglichen, sind jedoch meist erst in fortgeschrittenen Stadien sichtbar, wenn bereits Atrophien im MRT erkennbar und Symptome einer Demenz vorhanden sind. Besonders betroffen sind im FDG-PET der Precuneus und der posteriore Gyrus cinguli.
Beim Amyloid-PET wird häufig das C-11 Pittsburgh Compound B eingesetzt. Zunehmend werden auch fluorierte Liganden wie 18F-Florbetaben (ein mit 18F markiertes Stilben-Derivat) oder 18F-Florbetapir verwendet, die gezielt an Beta-Amyloid-Fibrillen binden. Die Menge der Amyloidablagerungen korreliert nicht direkt mit dem Grad der kognitiven Einschränkung. Da der Tracer stark lipophil ist, reichert er sich bei gesunden Personen vorwiegend in der weißen Substanz an. Sobald die Tracerkonzentration in der grauen Substanz das Niveau der weißen Substanz erreicht, wird die PET als „amyloid-positiv“ bewertet. Das Amyloid-PET eignet sich besonders gut, um eine Alzheimer-Erkrankung als Ursache einer Demenz auszuschließen.
Es stehen auch Tracer zur Verfügung, die Tau-Ablagerungen abbilden können. Diese binden an das Tau-Protein und zeigen eine verstärkte Aktivität in den betroffenen Hirnregionen (Hippocampus, entorhinaler Kortex, temporaler und parietaler Kortex). Die Aktivität korreliert mit dem Ausmaß der kognitiven Beeinträchtigung. Die Tau-PET ist jedoch nicht spezifisch für Alzheimer, da Tau-Ablagerungen auch bei anderen Erkrankungen wie der progressiven supranukleären Blickparese (PSP) oder der chronischen traumatischen Enzephalopathie vorkommen.
Therapie
Bis heute (2026) ist die Alzheimer-Krankheit unheilbar. Daher bezieht sich die gängige Therapie auf die Linderung der Symptome. Einige Medikamente konnten in klinischen Studien eine Verbesserung der Symptomatik (besonders der Merkfähigkeit) zeigen, unter anderem:
Darüber hinaus sind eine Gruppe von monoklonalen Antikörpern im Einsatz bzw. in klinischer Entwicklung, die sich gezielt gegen Beta-Amyloid richten und seinen Abbau durch das Immunsystem induzieren. Sie können vor allem im Frühstadium der Erkrankung den Progress verzögern. Dazu zählen
- Aducanumab,
- Lecanemab und
- Donanemab.
Die Behandlung ist zum Teil mit schweren Nebenwirkungen (z.B. Hirnödem, Hirnblutungen, Eisenablagerung) verbunden. Diagnostik (PET-CT, Biomarker im Liquor), Therapie und Nachuntersuchungen sind kostenintensiv. Die Langzeit-Prognose des Morbus Alzheimer wird durch die bislang entwickelten Wirkstoffe nicht signifikant verändert.[12]
Quellen
- ↑ Höfler et al. Lehrbuch Pathologie, 6. Auflage, 2019
- ↑ Banerjee, G., Farmer, S.F., Hyare, H. et al. Iatrogenic Alzheimer’s disease in recipients of cadaveric pituitary-derived growth hormone. Nat Med (2024).
- ↑ Gelbe Liste - Alzheimer: ApoE2 schlägt ApoE4, abgerufen am 28.03.2023
- ↑ Raulin et al. ApoE in Alzheimer’s disease: pathophysiology and therapeutic strategies. Molecular Neurodegeneration volume, 2022
- ↑ Medizinisch Genetisches Zentrum - AlzheimerTyp, familiär, abgerufen am 27.03.2023
- ↑ Deutsche Alzheimer Gesellschaft - Die Genetik der Alzheimer-Krankheit, abgerufen am 27.03.2023
- ↑ Alzheimer-Forschung - Ist Alzheimer erblich?, abgerufen am 27.03.2023
- ↑ Rollwagen. Einfluss systemischer Infektionen auf den Krankheitsverlauf der Alzheimer-Erkrankung im Maus-Modell, Inaugural-Dissertation zur Erlangung des Doktorgrades der Medizinischen Fakultät der Georg-August-Universität zu Göttingen, 2010
- ↑ Hope Y Agbemenyah et al.: Insulin growth factor binding protein 7 is a novel target to treat dementia, abgerufen am 11.1.2021
- ↑ Ferreira D et al. Biological subtypes of Alzheimer disease: A systematic review and meta-analysis. Neurology. 2020
- ↑ Josephs KA et al. Rates of hippocampal atrophy and presence of post-mortem TDP-43 in patients with Alzheimer's disease: a longitudinal retrospective study. Lancet Neurol. 2017
- ↑ Diener HC, Dodel R. Monoklonale Antikörper in der Therapie der Alzheimer-Erkrankung. Arzneimitteltherapie 2024
Literatur
- Finckh, Ulrich Genetische Faktoren bei Alzheimer Demenz / The role of genetics in Alzheimer disease Arztebl 2006;
- Thomas D Bird. et al. Alzheimer Disease Overview, Gene Reviews, 1998