Ehlers-Danlos-Syndrom









Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
Loslegennach den Dermatologen Edvard Ehlers (1863-1937) und Henry Alexandré Danlos (1844-1912)
Englisch: Ehlers-Danlos syndrome
Definition
Unter dem Ehlers-Danlos-Syndrom, kurz EDS, versteht man eine heterogene Gruppe von Bindegewebsstörungen, die z.B. durch Hypermobilität der Gelenke, eine überdehnbare Haut und instabile Gewebe gekennzeichnet sind.
Epidemiologie
Beim Ehlers-Danlos-Syndrom handelt es sich um eine seltene Erkrankung. Man schätzt die Häufigkeit auf 1:5.000 bis 1:10.000 ein. Das Ehlers-Danlos-Syndrom tritt bei beiden Geschlechtern in etwa gleich häufig auf.
Ätiopathogenese
Die Erkrankung beruht auf verschiedenen genetischen Defekten, die sich u.a. auf die Kollagensynthese auswirken. Nach der EDS-Klassifikation von 2017 werden 13 klinisch, genetisch und molekularbiologisch unterschiedliche Formen unterschieden (siehe unten).[1] Die Subtypen cEDS, cIEDS und cvEDS machen mehr als 80 % aller Fälle aus. Nur wenige Familien sind von den Subtypen vEDS, aEDS, dEDS und kEDS (früher Typ IV bis VII) betroffen. In Einzelfällen kommt es zum Auftreten der anderen Subtypen.
Bei den verschiedenen Formen des Ehlers-Danlos-Syndroms konnten Defekte verschiedener Gene nachgewiesen werden. Betroffen sind Gene, die für Faserproteine kodieren (z.B. COL1A1, COL1A2 und TNXB), oder Enzym-kodierende Gene (z.B. ADAMTS2 und PLOD1). Mutationen dieser Gene verändern die Synthese und Verarbeitung des Kollagens oder von Proteinen, die mit Kollagen interagieren. Sie führen so zu klinisch relevanten Strukturveränderungen.
Der Vererbungsmodus hängt vom genauen Typ des EDS ab. Die meisten Formen werden autosomal-dominant vererbt, d.h. nur eins der Allele muss verändert sein, um die Krankheit zum Ausbruch zu bringen. Einige Formen werden jedoch auch autosomal-rezessiv vererbt. Hier müssen beide Allele verändert sein, damit ein EDS in Erscheinung tritt.
Einteilung
EDS-Klassifikation von 2017
2017 wurden die Diagnosekriterien des Ehlers-Danlos-Syndroms umfassend überarbeitet und 13 Subtypen definiert. Die EDS-Klassifikation ersetzt die veraltete Villefranche-Klassifikation (s.u.).[1] Folgende Subtypen werden unterschieden:
| Subtyp | Synonyme | Erbgang | Protein (Gen) |
|---|---|---|---|
| Klassisches EDS | cEDS, früher Typ I | autosomal-dominant | Kollagen V (COL5A1), seltener Kollagen I ( COL1A1) |
| Classical-like EDS | clEDS, früher Typ II | autosomal-rezessiv | Tenascin X (TNXB) |
| Kardiovalvuläres EDS | cvEDS | autosomal-rezessiv | Kollagen I (COL1A2) |
| Vaskuläres EDS | vEDS, früher Typ IV | autosomal-dominant | Kollagen III (COL3A1), seltener Kollagen I (COL1A1) |
| Hypermobiles EDS | hEDS, früher Typ III | autosomal-dominant | unbekannt |
| Arthrochalasisches EDS | aEDS, früher Typ VII A/B | autosomal-dominant | Kollagen I (COL1A1, COL1A2) |
| Dermatosparaktisches EDS | dEDS, früher Typ VII C | autosomal-rezessiv | P1NP (ADAMTS2) |
| Kyphoskoliotisches EDS | kEDS, früher Typ VI | autosomal-rezessiv | PLOD1 (PLOD1), FKBP22 (FKBP14) |
| Brittle-Cornea-Syndrom | BCS | autosomal-rezessiv | Zinkfingerprotein 469 (ZNF469), PRDM5 (PRDM5) |
| Spondylodysplastisches EDS | spEDS | autosomal-rezessiv | β1,4-Galactosyltransferase 6 (B4GALT6), β1,4-Galactosyltransferase7 (B4GALT7), ZIP13 (SLC39A13) |
| Muskulokontrakturelles EDS | mcEDS | autosomal-rezessiv | Dermatan-4O-Sulfotransferase-1 (CHST14), Dermatansulfat-Epimerase (DSE) |
| Myopathisches EDS | mEDS | autosomal-rezessiv oder -dominant | Kollagen XII (COL12A1) |
| Parodontales EDS | pEDS | autosomal-dominant | C1s (C1S) und C1r (C1R) |
Villefranche-Klassifikation von 1997
Die klassische Villefranche-Klassifikation ist nicht mehr aktuell, wird teilweise aber noch verwendet. Sie unterscheidet 6 Haupttypen des EDS:[2]
- Klassischer Typ (Typ I und II): Autosomal-dominant vererbliche Gendefekte von COL5A1 oder COL5A2. Charakteristisch sind die stark überdehnbare, leicht verletzbare Haut. Die Gelenke sind überbeweglich, aber nicht so ausgeprägt wie beim hypermobilen Typ. Der Bewegungsapparat fällt durch Muskelhypotonie und Subluxation auf. Es besteht Blutungsneigung.
- Hypermobiler Typ (Typ III): Wird meist autosomal-dominant vererbt. Ursächlich ist in etwa 10 % der Fälle eine Mutation im TNXB-Gen. Die Haut ist nur gering beteiligt, es fällt aber eine ausgeprägte Hypermobilität der Gelenke auf. Daher haben viele Patienten aufgrund der unphysiologischen Beanspruchung der Gelenke chronische Schmerzen und Arthrosen. Neben der Hämatomneigung ist eine leicht verlängerte Blutungszeit auffällig.
- Vaskulärer Typ (Typ IV): Gendefekt von COL3A1 mit autosomal-dominantem Erbgang, bei dem die Gefäßproblematik am stärksten ausgeprägt ist. Bei diesen Patienten kommen häufig lebensbedrohliche Komplikationen wie Organrupturen und Aneursymen vor.
- Kyphoskoliotischer Typ (Typ VI): Gendefekt von PLOD1 mit Lysylhydroxylasemangel. Die Mutation wird autosomal-rezessiv vererbt. Schon bei Geburt bestehen eine starke Muskelhypotonie und eine progressive Skoliose. Die Gelenke sind hypermobil, die Haut ist brüchig und neigt zu atrophischen Narben. Am Auge kann eine Mikrokornea vorliegen.
- Arthrochalasischer Typ (Typ VII A/B): Bei diesem Typ liegt eine Mutation auf dem Exon 6 von COL1A1 oder COL1A2 vor. Die Mutation ist autosomal-dominant vererblich und führt zu einer Störung des Prokollagens vom Typ I. Es besteht eine starke Hypermobillität der Gelenke mit häufigen Subluxationen. Kongenital liegt oft eine bilaterale Hüftluxation vor. Weiterhin fallen Muskelhypotonie und Kyphoskoliose auf. Die Haut zeigt atrophische Narben und Hautbrüchigkeit. Wie bei anderen Formen häufig Blutungsneigung.
- Dermatosparaktischer Typ (Typ VII C): Diese autosomal-dominant vererbte Form beruht auf einem Gendefekt von ADAMTS2 mit konsekutivem Mangel an P1NP. Klinisch besteht eine starke Hautbrüchigkeit mit schlaffer und teigiger Haut. Hämatologisch kommt es zur Blutungsneigung. Die Patienten sind von kurzer Statur, ihre Augen haben eine blaue Sklera.
Klinik
Die bindegewebsreichen Strukturen von Haut, Blutgefäßen und Gelenken sind aufgrund der Störung der Kollagensynthese ungenügend ausgebildet, was zu einer fehlenden Festigkeit, einer Überdehnbarkeit des Bindegewebes und zu einem leichten Zerreißen der betroffenen Strukturen, insbesondere der Blutgefäße, führen kann. Auch Darmrupturen, Hernien, Verkrümmungen der Wirbelsäule und rezidivierende Pneumothoraces sind möglich.
Haut
Bei Patienten, die vom Ehlers-Danlos-Syndrom betroffen sind, kann die Haut am Hals, im Gesicht und an den Gelenken bis zu mehreren Zentimetern abgehoben werden und schnellt nach Loslassen wieder zurück, was auch als Cutis hyperelastica bezeichnet wird. Weiterhin kommt es zu einer verzögerten Wundheilung, wobei Wundränder auseinander klaffen und atrophische, minderwertige Narben entstehen. Die Ausbildung von hypertrophischen Narbenarealen, die über keinerlei Festigkeit verfügen, ist möglich.
Gelenke
An den Gelenken zeigt sich eine Überbeweglichkeit ohne Festigkeit. Die Gelenke können überstreckt werden und andere bizarre Bewegungen ausführen, was auch zu dem Begriff Schlangenmenschen geführt hat. Luxationen und Fehlstellungen sind häufig, gelegentlich auch im Bereich der Wirbelsäule (z.B. atlantoaxiale Subluxation).
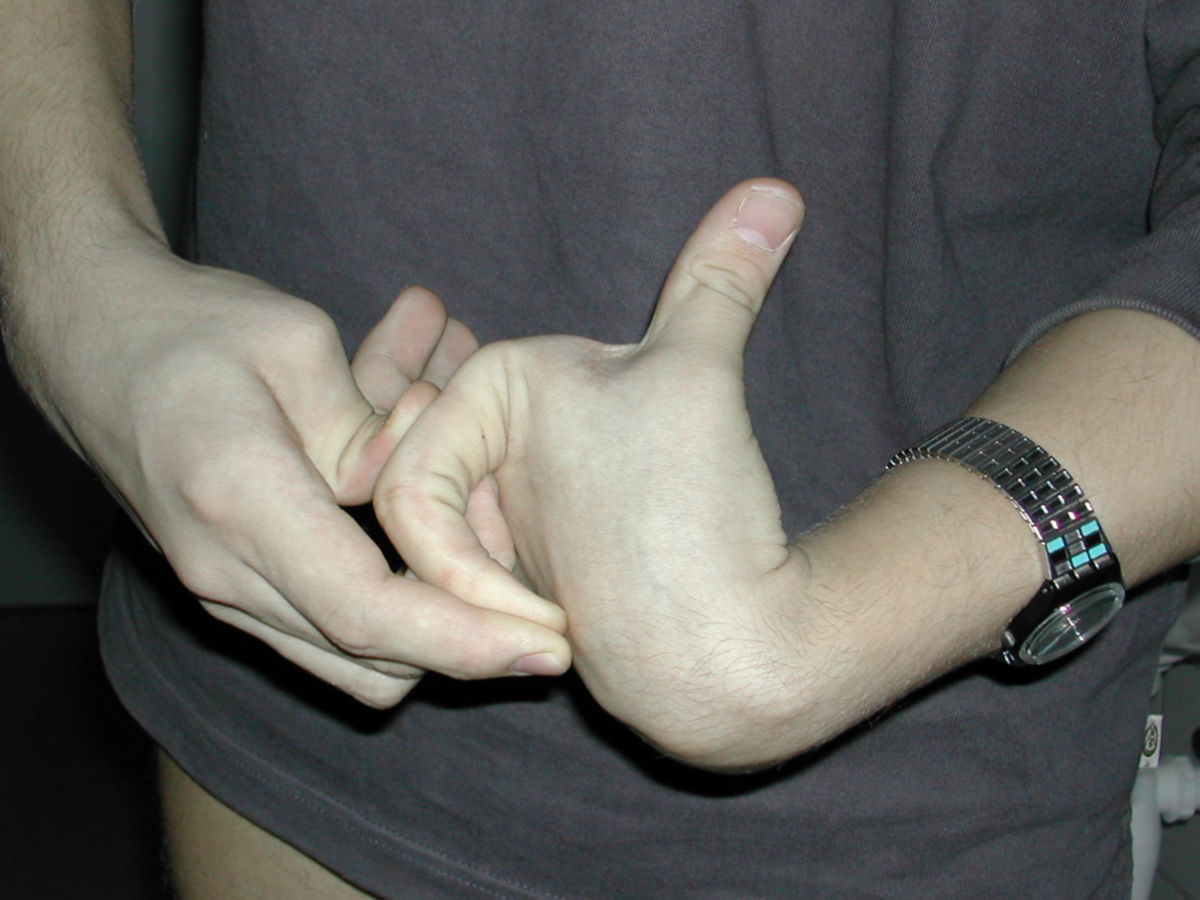
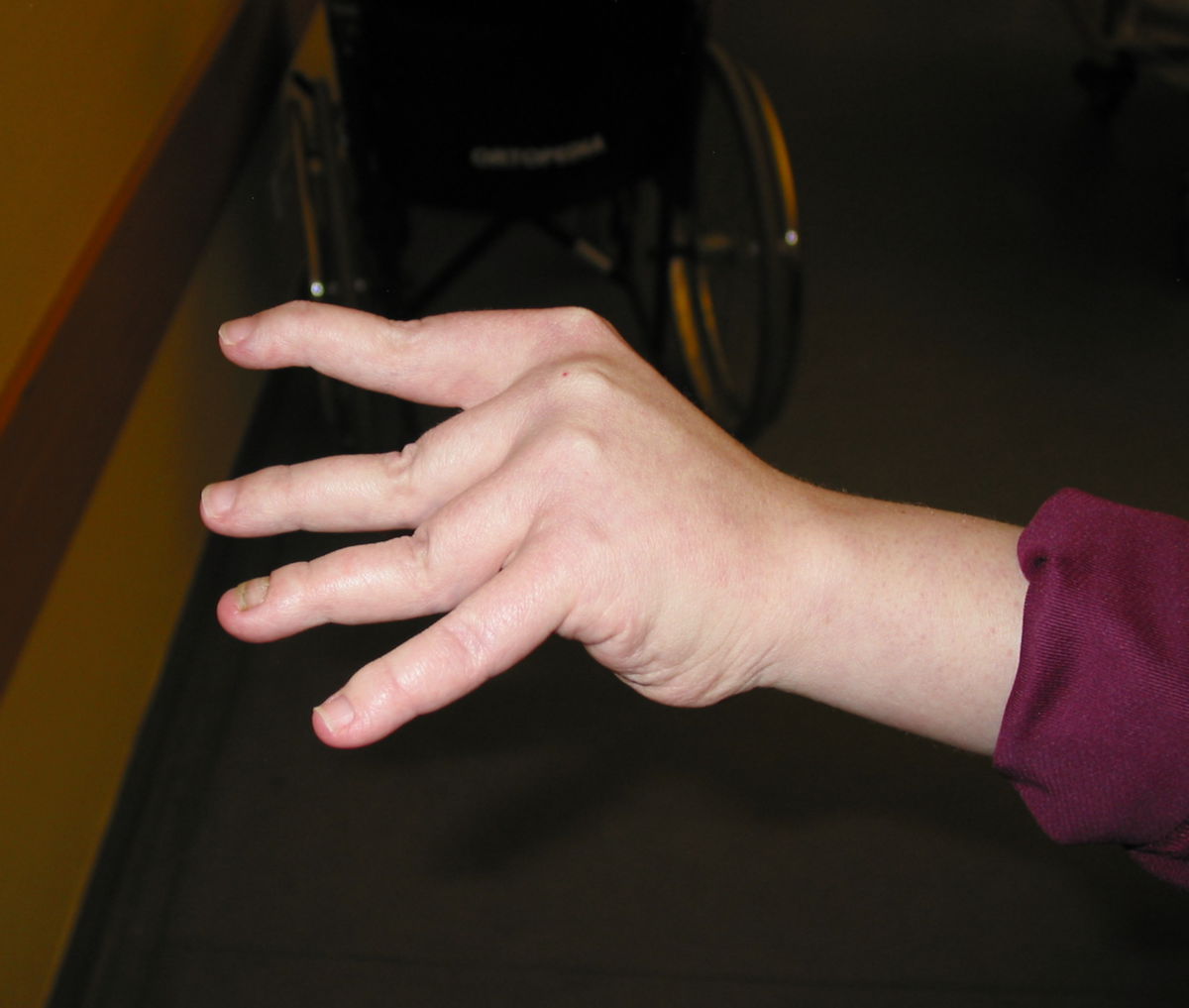
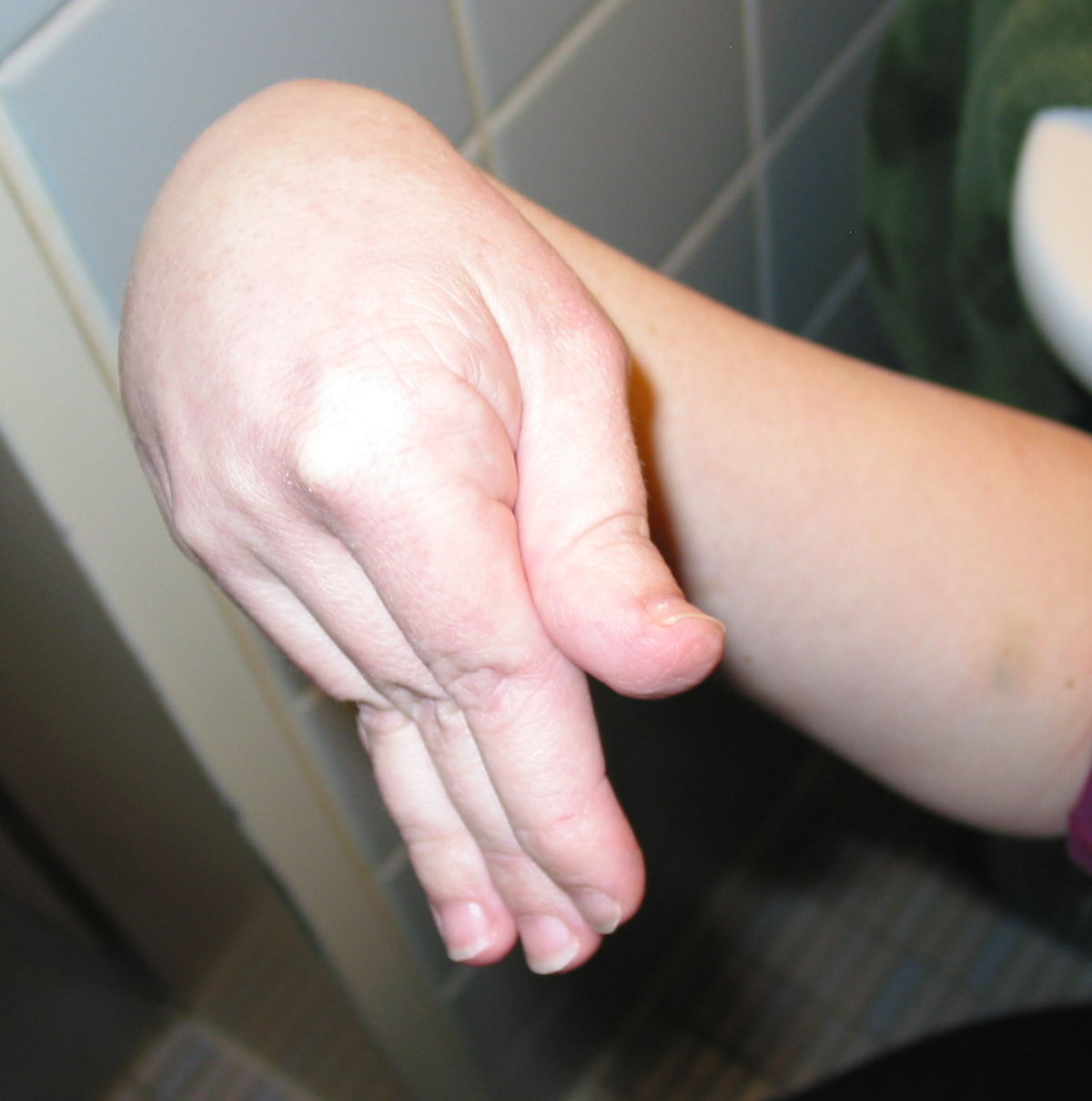
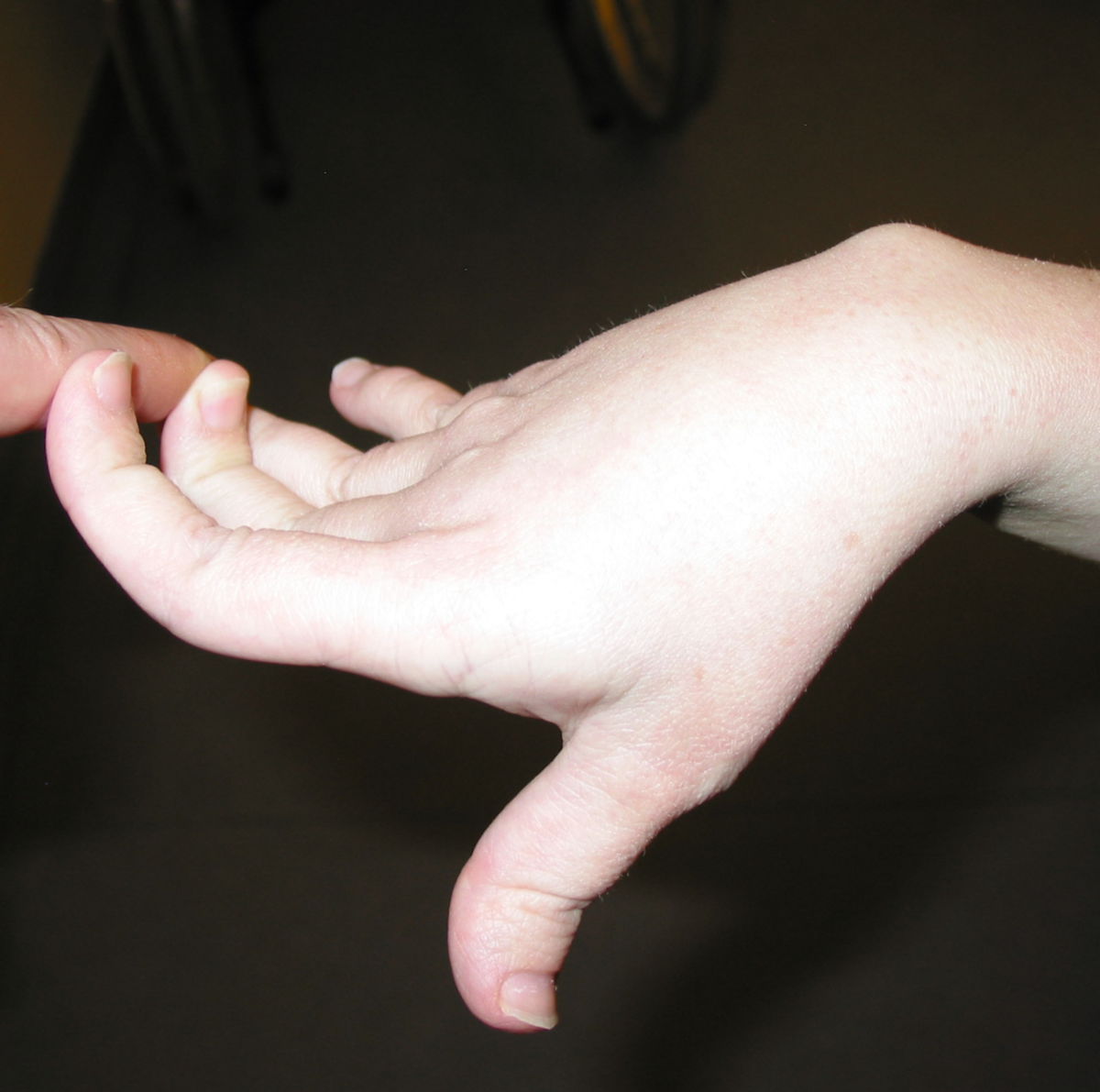
Gefäße
Die leichte Zerreißbarkeit von Gefäßen manifestiert sich an kleineren Gefäßen durch Ekchymosen und an größeren Gefäßen durch ausgeprägte Blutungen, die vor allem durch Schwangerschaft und Geburt sowie sportliche Betätigung und Traumen ausgelöst werden.
Diagnostik
Die Diagnose wird meist klinisch gestellt, wobei die positive Familienanamnese wegweisend ist. Als weitere diagnostische Maßnahmen kommen in Betracht:
- Beighton-Score zur Abklärung der Überbeweglichkeit
- Darstellung der erhöhten Kapillarfragilität kann durch einen Rumpel-Leede-Test
- Hautbiopsie mit anschließender elektronenmikroskopischer Untersuchung der Kollagenstruktur
Die Differenzierung in die verschiedenen EDS-Typen erfolgt molekularbiologisch mittels DNA-Amplifikation und anschließender Sequenzanalyse der DNA mit Nachweis von Mutationen der relevanten Gene.
Therapie
Aktuell (2024) existiert keine kausale Therapie. Die Behandlung ist daher rein symptomatisch und präventiv. Den betroffenen Patienten wird empfohlen, auf ausgeprägte Belastungen der Gelenke zu verzichten. Da die Wundheilung gestört ist, kommt der Prävention von Traumata eine wichtige Rolle zu. Ebenso sind Operationsindikationen zurückhaltend zu stellen. Schwangerschaften können das Blutungsrisiko erhöhen. Patienten sollten daher eine ausführliche Beratung in Anspruch nehmen und über die Risiken aufgeklärt werden.
Prognose
Es handelt sich um eine chronische Erkrankung, die progredient verläuft. Die Lebensqualität der betroffenen Patienten ist häufig eingeschränkt.
Weblinks
- Ehlers–Danlos Society
- National Institute of Health on Hypermobility EDS
- Ehlers–Danlos Syndrome, Classic Type (inkl. Ehlers–Danlos Syndrom Typ I & II)
- GeneReviews/NCBI/NIH/UW (Eintrag zu Ehlers–Danlos Syndrom Typ IV)
- Management of pain and fatigue in the joint hypermobility syndrome (a.k.a. Ehlers-Danlos syndrome, hypermobility type): principles and proposal for a multidisciplinary approach In: Am J Med Genet A. 2012 Aug; 158A(8):2055-70. doi: 10.1002/ajmg.a.35483.
- University of Washington Medicine, Orthopaedics and Sports Medicine (inkl. Symptome, Genetik, Diagnostik und Therapie)
Einzelnachweis
- ↑ 1,0 1,1 Malfait et al. The 2017 international classification of the Ehlers–Danlos syndromes American Journal of Medical Genetics, 2017
- ↑ Beighton et al.: "Ehlers-Danlos syndromes: Revised nosology, Villefranche, 1997" American Journal of Medical Genetics, 1999.