Polycythaemia vera










Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenSynonyme: PCV, Vaquez-Osler-Krankheit, Morbus Vaquez-Osler, Polycythaemia rubra vera
Englisch: polycythemia vera
Definition
Bei der Polycythaemia vera, kurz PV, handelt es sich um eine seltene myeloproliferative Erkrankung, bei der es zu einer autonomen Proliferation aller drei myeloischen Zellreihen im Knochenmark kommt. Davon sind insbesondere Erythrozyten betroffen, jedoch auch Thrombozyten und Leukozyten. Auslöser ist die Mutation einer hämatopoetischen Stammzelle.
siehe auch: Myelopoese
Epidemiologie
Polycythaemia vera ist eine seltene Erkrankung. Die Inzidenz wird international mit 0,4–2,8 Erkrankungen pro 100.000 Einwohner pro Jahr angegeben; für Deutschland ergab eine aktuelle Analyse von Krankenkassendaten eine Prävalenz von 28,6 und eine Inzidenz von 3,3 pro 100.000 Erwachsenen (Datenjahr 2021).[1] Das mediane Alter bei Diagnosestellung liegt bei etwa 65 Jahren. Männer sind häufiger betroffen als Frauen.
Pathogenese
Die Ätiologie der Erkrankung ist nicht geklärt. Man vermutet u.a. die Beteiligung ionisierender Strahlung und chemischer Noxen (z.B. Benzol) als Auslöser der Erkrankung. Meist ist jedoch kein Auslöser eruierbar.
In mehr als 95 % der Fälle liegt eine Mutation des Gens für die Januskinase 2 (JAK2) vor, überwiegend eine JAK2-V617F-Punktmutation (ca. 95 %), in einem kleineren Teil der Fälle eine JAK2-Exon-12-Mutation (ca. 2–3 %). In ca. 20 % der Fälle entwickelt sich im Krankheitsverlauf eine Post-PV-Myelofibrose (sekundäre Myelofibrose) – abzugrenzen von der primären Myelofibrose, die de novo ohne vorausgehende PV auftritt.
Klinik
Im Vordergrund steht eine Polyglobulie mit Erythrozytose. Das Blutvolumen ist insgesamt meist stark vermehrt.
Die Vermehrung der Erythrozyten macht sich anfangs als Hautrötung (Plethora) bemerkbar, im weiteren Verlauf auch als anfallsweise auftretende Erythromelalgie. Später, bei Hämatokrit-Werten über 55 %, kann es zum Hyperviskositätssyndrom kommen, da die Zähigkeit des Blutes (Blutviskosität) deutlich ansteigt.
Eine besonders gefährliche Folge davon ist eine Mangeldurchblutung des Gehirns (von leichtem Schwindel, TIA bis hin zum Schlaganfall), Mangeldurchblutung der Herzkranzgefäße (Angina pectoris bis Myokardinfarkt), sowie periphere Durchblutungsstörungen (Akrozyanose). Es kann eine Hypertonie entstehen.
Im fortschreitenden Krankheitsverlauf kann es zu Splenomegalie (ca. 75 %), Hepatomegalie (ca. 30 %) und – ab einem Hämatokrit von ca. 60 % – zu gefährlichen Thrombosen bzw. Thromboembolien (tiefe Beinvenenthrombosen, Lungenembolie etc.) kommen. Häufig ist aber auch eine verstärkte Blutungsneigung (hämorrhagische Diathese) zu beobachten, die trotz Thrombozytose aufgrund der Thrombopathie auftritt. Zusätzlich quält die Patienten zum Teil ein starker Pruritus, der oft nicht auf Therapie anspricht.
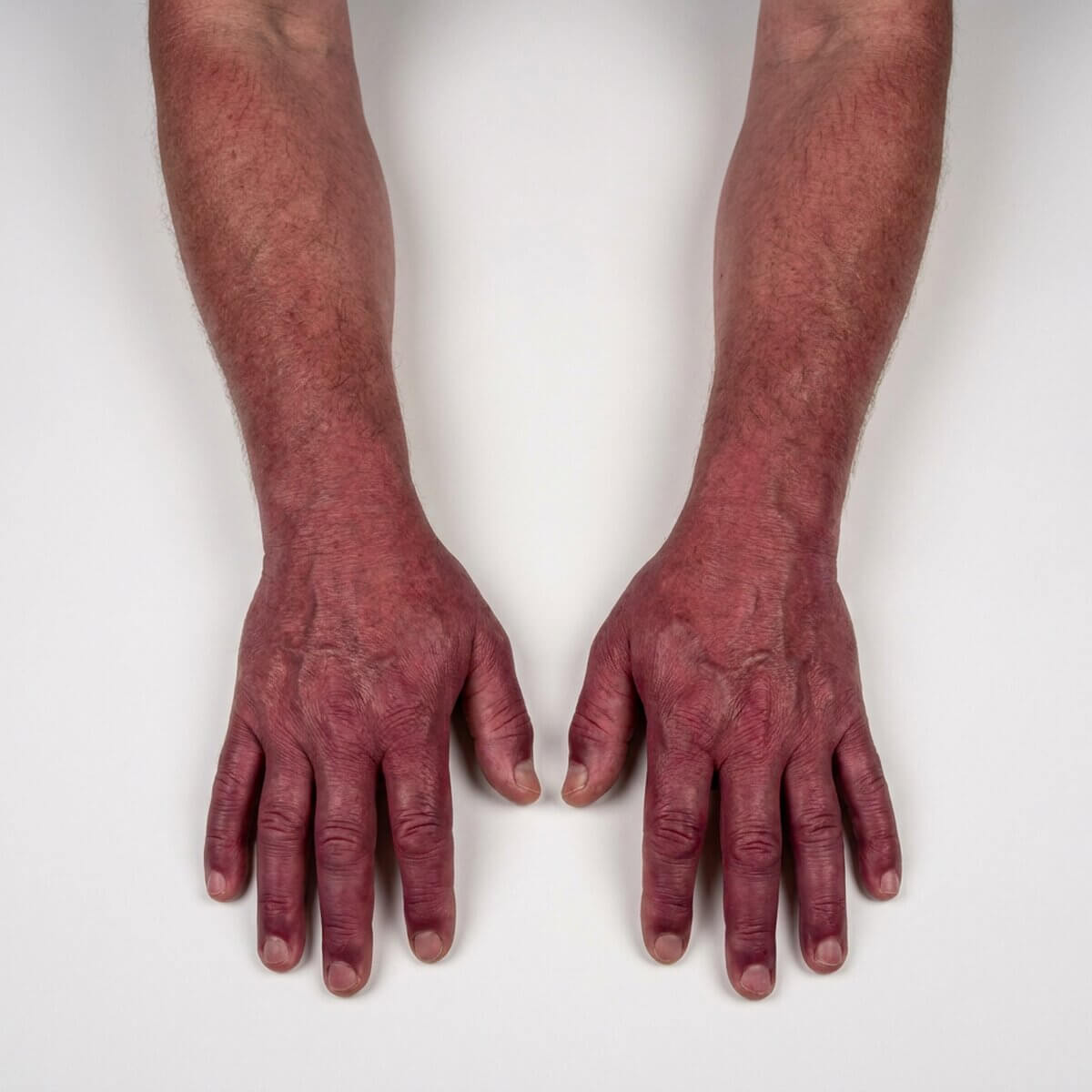
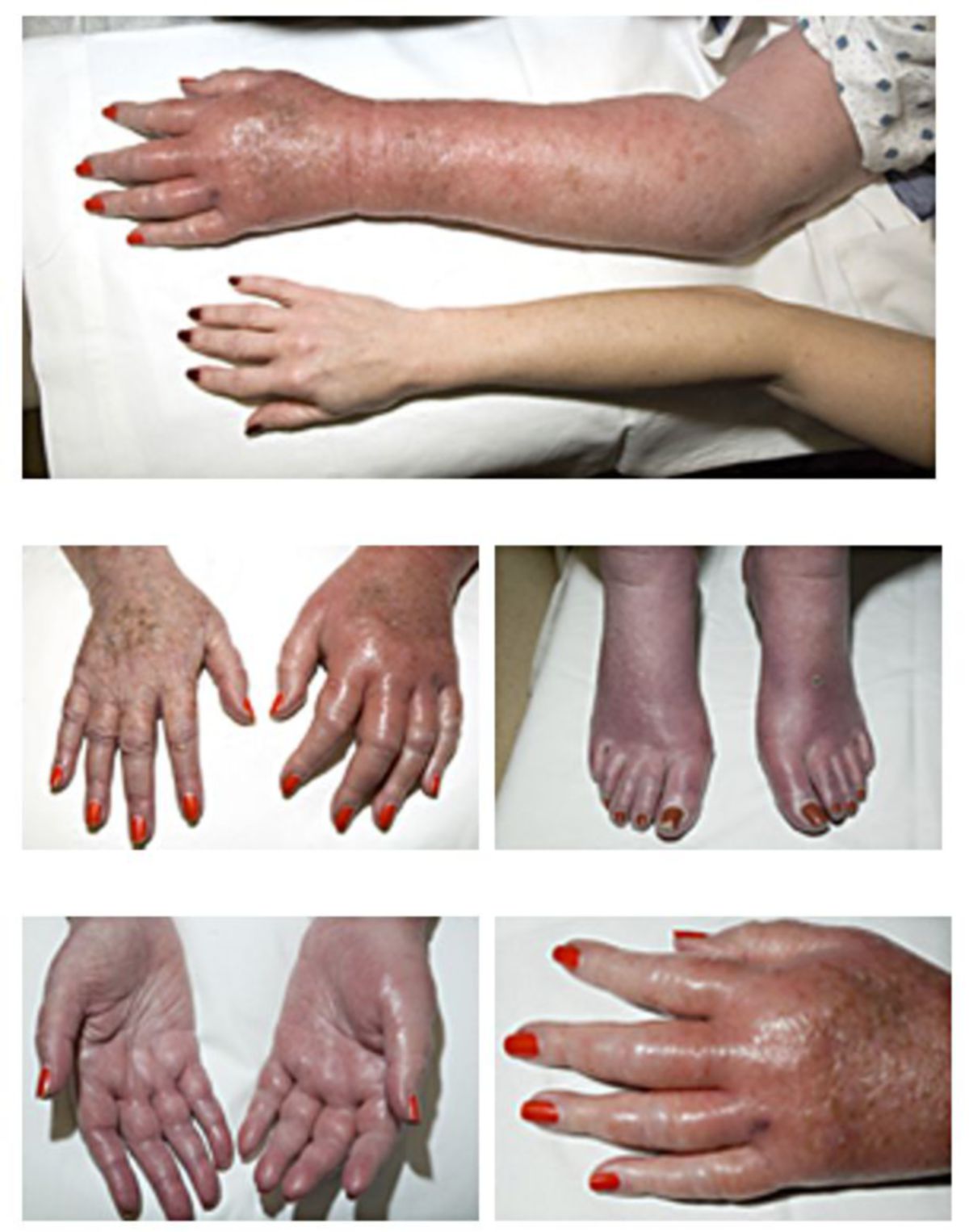
Diagnostik
Die Labordiagnostik zeigt folgende Befunde:
- erhöhter Hämatokrit und Hämoglobingehalt
- Erythrozytose, Leukozytose (mit relativer Lymphopenie) und Thrombozytose
- erniedrigtes Erythropoetin
- Basophilie
- Eosinophilie
- erhöhte alkalische Leukozytenphosphatase (ALP)
- erhöhte LDH sowie Harnsäure (vermehrter Zellzerfall)
- erniedrigte BSG
- erniedrigtes Serumeisen
- erhöhtes Bilirubin im Serum
- normwertiger pO2 (zur Abgrenzung einer kompensatorischen Polyglobulie)
Die Pathohistologie der Knochenmarkpunktion zeigt ein hyperzelluläres Knochenmark und eine Eisenverarmung. In über 90 % der Fälle lässt sich durch eine molekulargenetische Untersuchung eine JAK2-Mutation nachweisen.
Diagnosekriterien
Etabliert sind die WHO-Diagnosekriterien der 5. Auflage (2022). Sie entsprechen weitgehend den Kriterien der International Consensus Classification (ICC) 2022.[2]
Eine PV wird diagnostiziert, wenn alle drei Hauptkriterien oder die ersten beiden Hauptkriterien plus das Nebenkriterium erfüllt sind.
| Kriterium | Beschreibung |
|---|---|
| Hauptkriterien | |
| 1 | Hämoglobin > 16,5 g/dl (Männer) bzw. > 16,0 g/dl (Frauen) oder Hämatokrit > 49 % (Männer) bzw. > 48 % (Frauen) |
| 2 | Knochenmarkbiopsie mit altersbezogener Hyperzellularität und trilineärer Proliferation (Panmyelose), insbesondere ausgeprägte erythroide, granulozytäre und megakaryozytäre Proliferation mit pleomorphen, reifen Megakaryozyten |
| 3 | Nachweis einer JAK2-V617F- oder JAK2-Exon-12-Mutation |
| Nebenkriterium | |
| / | Erniedrigter Serum-Erythropoetin-Spiegel |
Hinweis: Referenzwerte sind häufig vom Messverfahren abhängig und können von den o.a. Werten abweichen. Ausschlaggebend sind die Referenzwerte, die vom Labor angegeben werden, das die Untersuchung durchführt.
Kriterium 2 (Knochenmarkbiopsie) kann entfallen, wenn eine anhaltende absolute Erythrozytose vorliegt (Hb > 18,5 g/dl bzw. Hkt > 55,5 % bei Männern, Hb > 16,5 g/dl bzw. Hkt > 49,5 % bei Frauen) und gleichzeitig Kriterium 3 sowie das Nebenkriterium erfüllt sind.
Merke: Die früher gebräuchlichen PVSG-Kriterien (Hämatokrit-Schwelle "> 25 % über Normwert", Kategorie-A/B-System mit Splenomegalie, Thrombozytose, Leukozytose und in-vitro-Koloniebildung als Diagnosekriterien) sind obsolet und wurden bereits durch die WHO-Kriterien 2008 bzw. 2016 abgelöst.
Risikostratifizierung
Die Risikostratifizierung ist entscheidend für die individualisierte Therapieplanung. Patienten werden in zwei Risikogruppen eingeteilt:
- Niedrigrisiko: Alter < 60 Jahre und keine thromboembolischen Ereignisse in der Anamnese
- Hochrisiko: Alter ≥ 60 Jahre und/oder thromboembolische Ereignisse in der Anamnese
Zusätzliche Faktoren wie kardiovaskuläre Risikofaktoren, extreme Thrombozytose (> 1.500 × 109/l), ausgeprägte Symptomlast oder Unverträglichkeit der Aderlasstherapie können auch bei formal niedrigem Risiko den Einsatz einer zytoreduktiven Therapie rechtfertigen.[3] Unter zytoreduktiver Therapie gilt zudem eine Senkung der JAK2-V617F-Allellast als prognostisch günstig.[3]
Therapie
Eine kurative Behandlung ist nur durch eine allogene Stammzelltransplantation möglich, die jedoch aufgrund der hohen Risiken nur bei ausgewählten jüngeren Hochrisikopatienten erwogen wird. In der Praxis erfolgt die Therapie meist symptomorientiert:
- Aderlass (Ziel-Hämatokrit < 45 %, geschlechtsunabhängig)[3]
- Thrombozytenaggregationshemmer (z.B. Acetylsalicylsäure 100 mg/Tag), sofern keine Kontraindikation besteht
Hinweis: Diese Dosierungsangaben können Fehler enthalten. Ausschlaggebend ist die Dosierungsempfehlung in der Herstellerinformation.
- Allopurinol bei Hyperurikämie zur Gichtprophylaxe
Reicht die Hämatokritkontrolle durch Aderlässe (alle 4–8 Wochen) nicht aus oder liegen Hochrisikofaktoren bzw. die oben genannten Zusatzkriterien vor, ist eine zytoreduktive Therapie indiziert. Alle Patienten, die eine Zytoreduktion benötigen und dafür infrage kommen, werden leitliniengerecht in der Erstlinie mit Interferon-alpha behandelt werden:[3]
- Interferon-alpha, bevorzugt Ropeginterferon alfa-2b (pegyliertes Interferon mit längerer Halbwertszeit; Gabe alle zwei Wochen) – Mittel der Wahl unabhängig von Alter und Thromboserisiko, sofern keine Kontraindikationen (z.B. symptomatische Splenomegalie) vorliegen
- Hydroxyharnstoff – Alternative, wenn Interferon-alpha nicht infrage kommt oder nicht vertragen wird
Bei Resistenz oder Unverträglichkeit gegenüber Erstlinientherapien kann eine Eskalation erfolgen mit
- Ruxolitinib (JAK-Inhibitor)
- Busulfan
- Radiophosphor (P32) – heute wegen des erhöhten leukämogenen Risikos nur noch in Ausnahmefällen bei älteren Patienten
Komplikationen
Kritische Komplikationen sind:
- Thrombozytendysfunktion:
- thromboembolische Ereignisse (Myokardinfarkt, Hirninfarkt)
- Thrombosen (z.B. Budd-Chiari-Syndrom)
- Hämorrhagische Diathesen
- Übergang in andere myeloproliferative Erkrankungen:
Prognose
Betroffene können eine normale Lebenserwartung haben, wenn die Erkrankung engmaschig überwacht und leitliniengerecht behandelt wird. Die mediane Überlebenszeit liegt unter adäquater Therapie bei über 20 Jahren. Prognostisch ungünstig sind ein höheres Lebensalter bei Diagnosestellung, thromboembolische Ereignisse in der Anamnese sowie eine ausgeprägte Leukozytose. Unbehandelt beträgt die mittlere Überlebenszeit bei Polycythaemia vera aufgrund der vaskulären Komplikationen nur wenige Jahre.
Quellen
- ↑ Manz KC, Mocek A, Morouj B et al. Prevalence, incidence, and thromboembolic events in polycythemia vera: a study based on longitudinal German health claims data. Ann Hematol. 2025;104(1):347-360.
- ↑ Khoury et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022.
- ↑ 3,0 3,1 3,2 3,3 Lengfelder E, Baerlocher GM, Döhner K et al. Polycythaemia Vera (PV). Onkopedia-Leitlinie. Deutsche Gesellschaft für Hämatologie und Onkologie e.V. (DGHO); Stand September 2023. Verfügbar unter: Onkopedia-Leitlinie Polycythaemia vera.