Galaktose








Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenSynonyme: Galactose, Cerebrose
Englisch: galactose
Definition
Galaktose, kurz Gal, ist ein natürlich vorkommendes Monosaccharid (Einfachzucker) aus der Gruppe der Hexosen mit der Summenformel C6H12O6. Sie schmeckt süß, jedoch deutlich schwächer als Glukose.[1]
Chemie
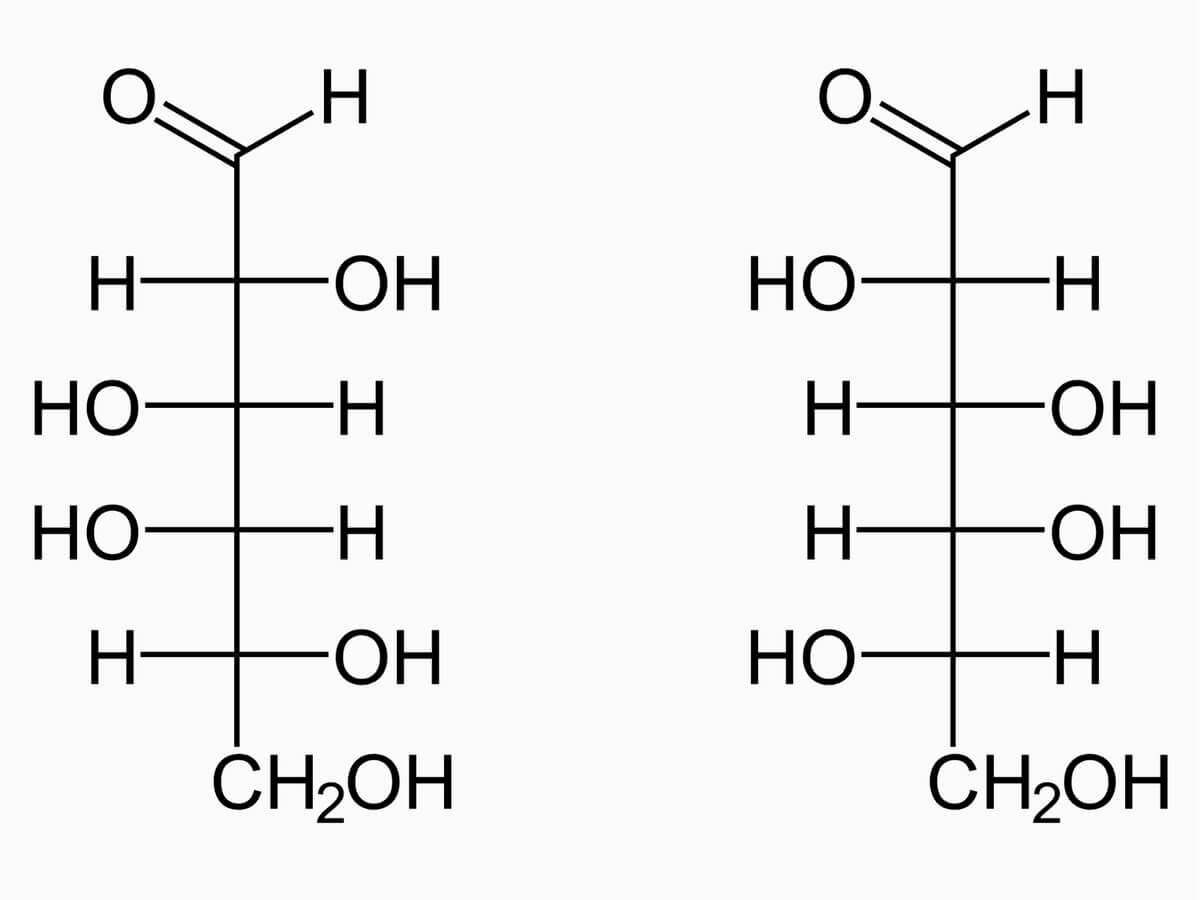
Galaktose weist sechs Kohlenstoffatome auf und gehört deshalb zu den Hexosen. Ihr C1-Atom trägt eine Aldehydgruppe, sodass es sich um eine Aldose handelt. Galaktose ist das C4-Epimer der Glukose und unterscheidet sich von dieser somit nur durch die Konfiguration der Hydroxylgruppe am C4-Atom. Physiologisch relevant ist das D-Enantiomer (D-Galaktose).
In wässriger Lösung bildet Galaktose ein Halbacetal aus: Das Carbonyl-C-Atom verbindet sich mit der Hydroxylgruppe des C5-Atoms unter Bildung eines Sechserringes (Pyranose). Dabei entstehen zwei Anomere (α- und β-D-Galaktopyranose), die in Lösung über die Mutarotation ineinander übergehen.
Vorkommen
Galaktose kommt in der Natur überwiegend gebunden vor. Wichtigste Quelle in der menschlichen Ernährung ist das Disaccharid Laktose (Milchzucker), das aus Galaktose und Glukose besteht und in der Milch von Säugetieren enthalten ist. Darüber hinaus ist Galaktose Bestandteil zahlreicher Glykolipide, Glykoproteine und Glykosaminoglykane.
Biochemie
Galaktose ist ein wichtiger Metabolit des menschlichen Organismus. Die über die Nahrung – vor allem nach enzymatischer Spaltung von Laktose – aufgenommene Galaktose wird im Darm resorbiert und in der Leber über den sogenannten Leloir-Weg in den Kohlenhydratstoffwechsel eingeschleust.[1] Dieser umfasst vier enzymatische Schritte:
- Zunächst wandelt die Galaktose-Mutarotase (GALM) die β-D-Galaktose in α-D-Galaktose um.
- Die Galaktokinase (GALK1) phosphoryliert die α-D-Galaktose unter ATP-Verbrauch am C1-Atom zu Galaktose-1-phosphat.
- Anschließend katalysiert die Galaktose-1-Phosphat-Uridyltransferase (GALT) die Übertragung einer Uridylgruppe von UDP-Glukose auf Galaktose-1-phosphat. Es entstehen Glukose-1-phosphat und UDP-Galaktose.
- Im letzten Schritt wird die UDP-Galaktose durch die UDP-Galaktose-4-Epimerase (GALE) am C4-Atom zu UDP-Glukose epimerisiert. Die Reaktion verläuft über ein NAD+-abhängiges Oxidations-Reduktions-Prinzip mit einem 4-Keto-Zwischenprodukt.
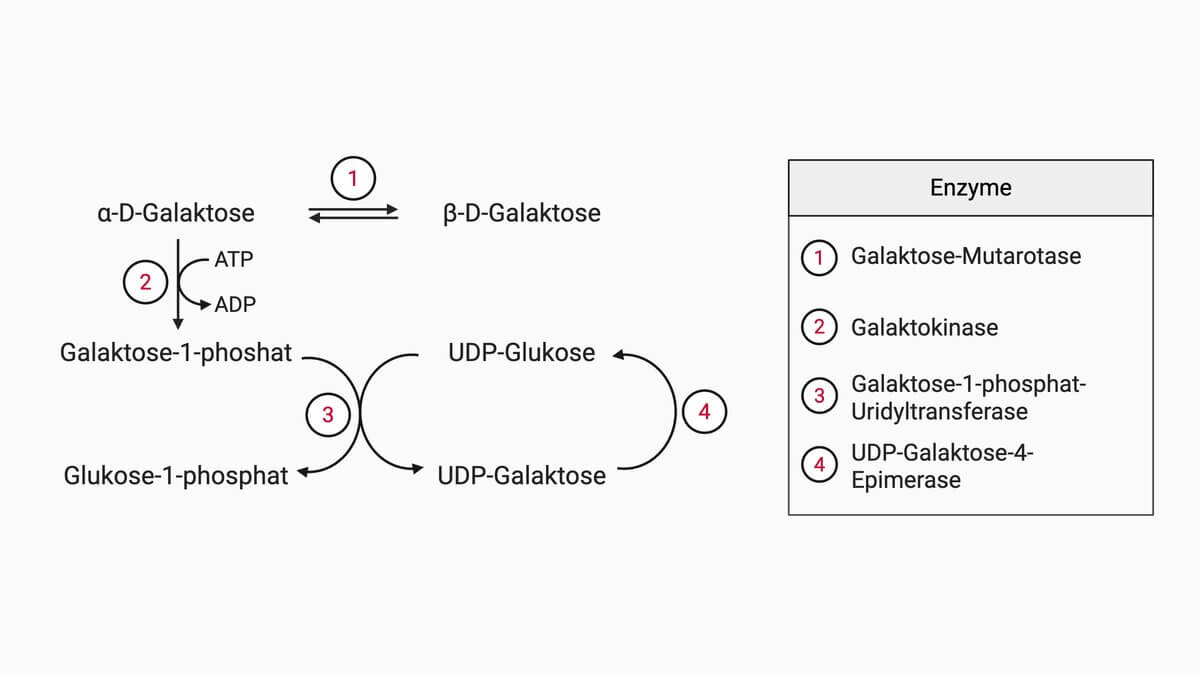
Die so gebildete UDP-Glukose kann zum Aufbau von Glykogen verwendet oder bei Bedarf in die katabolen Stoffwechselwege eingeführt werden. Da die GALE-Reaktion reversibel ist, kann Galaktose umgekehrt auch endogen aus Glukose synthetisiert werden – eine galaktosefreie Ernährung schließt eine Galaktoseexposition daher nie vollständig aus.[1]
Galaktose ist außerdem ein wichtiger Baustein verschiedener Membranlipide, insbesondere der Cerebroside und Ganglioside, sowie der Laktose, die in der laktierenden Brustdrüse aus UDP-Galaktose und Glukose synthetisiert wird.
Klinik
Bei einem Defekt der am Galaktosestoffwechsel beteiligten Enzyme kommt es zur Anreicherung von Galaktose und ihren Stoffwechselprodukten. Diese Stoffwechselstörungen werden unter dem Begriff Galaktosämie zusammengefasst. Insgesamt werden vier Formen unterschieden:[2][3]
- Typ 1: klassische Galaktosämie (Defekt der Galaktose-1-Phosphat-Uridyltransferase)
- Typ 2: Galaktokinasemangel
- Typ 3: Uridindiphosphat-Galaktose-4-Epimerase-Mangel
- Typ 4: Galaktose-Mutarotase-Mangel[4]
Die klassische Galaktosämie (Typ 1) manifestiert sich bereits im Neugeborenenalter und kann unbehandelt lebensbedrohlich verlaufen. Ursächlich ist insbesondere die toxische Akkumulation von Galaktose-1-phosphat. Über die Aldosereduktase entsteht zudem Galaktitol, dessen Anreicherung in der Augenlinse zur Ausbildung einer Katarakt beiträgt.[2] In Deutschland wird die klassische Galaktosämie im Rahmen des Neugeborenenscreenings erfasst.
Therapeutisch steht eine lebenslange laktose- und galaktosearme Ernährung im Vordergrund. Diese verhindert die akuten Komplikationen, kann jedoch Langzeitfolgen wie kognitive Beeinträchtigungen und eine Ovarialinsuffizienz nicht vollständig vermeiden.[1][2]
Quellen
- ↑ 1,0 1,1 1,2 1,3 Succoio M, Sacchettini R, Rossi A, Parenti G, Ruoppolo M. Galactosemia: Biochemistry, Molecular Genetics, Newborn Screening, and Treatment. Biomolecules. 2022;12(7):968.
- ↑ 2,0 2,1 2,2 Demirbas D, Coelho AI, Rubio-Gozalbo ME, Berry GT. Hereditary galactosemia. Metabolism. 2018;83:188-196.
- ↑ Wada Y, Kikuchi A, Arai-Ichinoi N, et al. Biallelic GALM pathogenic variants cause a novel type of galactosemia. Genet Med. 2019;21(6):1286-1294.
- ↑ Banford S, Timson DJ. The structural and molecular biology of type IV galactosemia. Biochimie. 2021;183:13-17.