UDP-Galaktose-4-Epimerase


Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenSynonym: UDP-Glukose-4-Epimerase
Englisch: UDP-galactose-4-epimerase, UDP-glucose-4-epimerase
Definition
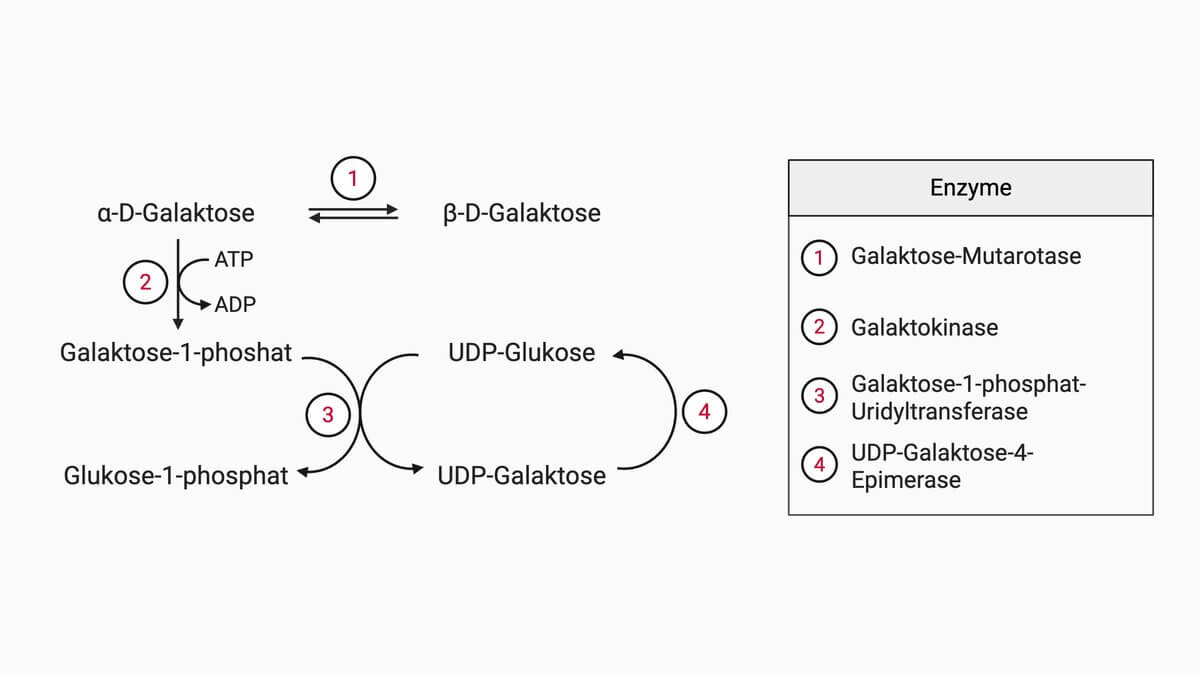
Die UDP-Galaktose-4-Epimerase, kurz GALE, ist ein Enzym des Galaktosestoffwechsels, das UDP-Galaktose und UDP-Glukose reversibel ineinander umwandelt. Sie gehört zur Enzymklasse der Isomerasen bzw. Epimerasen und katalysiert den vierten Schritt des Leloir-Wegs.[1]
Genetik
Die UDP-Galaktose-4-Epimerase wird durch das GALE-Gen kodiert, das auf dem kurzen Arm von Chromosom 1 (Genlokus 1p36) lokalisiert ist.
Biochemie
Die Epimerisierung erfolgt am C4-Atom des an UDP gebundenen Zuckerrests. Mechanistisch verläuft sie über ein fest gebundenes NAD+ als Cofaktor: Dieses oxidiert die Hydroxylgruppe am C4-Atom zu einer Ketogruppe (4-Keto-Zwischenprodukt), die anschließend von der gegenüberliegenden Seite wieder reduziert wird, wodurch die Konfiguration am C4-Atom umgekehrt wird. Es handelt sich somit um ein Oxidations-Reduktions-Prinzip. Das humane Enzym liegt als Homodimer vor.[1]
Da die Reaktion reversibel ist, kann der Organismus UDP-Galaktose auch endogen aus UDP-Glukose bilden und ist damit nicht zwingend auf eine exogene Galaktosezufuhr angewiesen. Über die zweite Teilreaktion stellt das Enzym zudem UDP-N-Acetylgalaktosamin bereit, das als Baustein für die Synthese von Glykoproteinen und Glykolipiden benötigt wird.
Reaktion
Das humane Enzym ist bifunktionell und katalysiert zwei analoge, reversible Reaktionen:
Klinik
Biallele Varianten im GALE-Gen führen zum UDP-Galaktose-4-Epimerase-Mangel, der als Galaktosämie Typ III (Epimerasemangel-Galaktosämie) bezeichnet wird. Das klinische Spektrum reicht von einer milden, peripheren Form (Enzymdefekt nur in Erythrozyten, klinisch meist unauffällig) bis zu einer schweren, generalisierten Form, die ähnlich wie die klassische Galaktosämie verläuft (u.a. Leberschädigung, früh auftretende Katarakt, Innenohrschwerhörigkeit und kognitive Beeinträchtigung).[2]
Aufgrund der bifunktionellen Enzymaktivität sind Betroffene der schweren Form nicht nur auf eine kontrollierte Galaktosezufuhr, sondern auch auf eine ausreichende exogene Zufuhr von N-Acetylgalaktosamin angewiesen, da dieser Baustein bei Enzymdefekt nicht ausreichend endogen bereitgestellt werden kann.[1]
Darüber hinaus wurden biallele GALE-Varianten mit einer syndromalen Thrombozytopenie (Thrombozytopenie Typ 13) in Verbindung gebracht.[3]
Quellen
- ↑ 1,0 1,1 1,2 Succoio M, Sacchettini R, Rossi A, Parenti G, Ruoppolo M. Galactosemia: Biochemistry, Molecular Genetics, Newborn Screening, and Treatment. Biomolecules. 2022;12(7):968.
- ↑ Demirbas D, Coelho AI, Rubio-Gozalbo ME, Berry GT. Hereditary galactosemia. Metabolism. 2018;83:188-196.
- ↑ GALE, OMIM, abgerufen am 18.06.2026