Gastrointestinaler Stromatumor










Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenEnglisch: gastrointestinal stromal tumor
Definition
Gastrointestinale Stromatumoren, kurz GIST, sind die häufigsten mesenchymalen Tumoren (Sarkome) des Gastrointestinaltraktes, die erst seit 1998 eindeutig diagnostiziert werden können. In den meisten Fällen treten GIST sporadisch auf.
Hintergrund
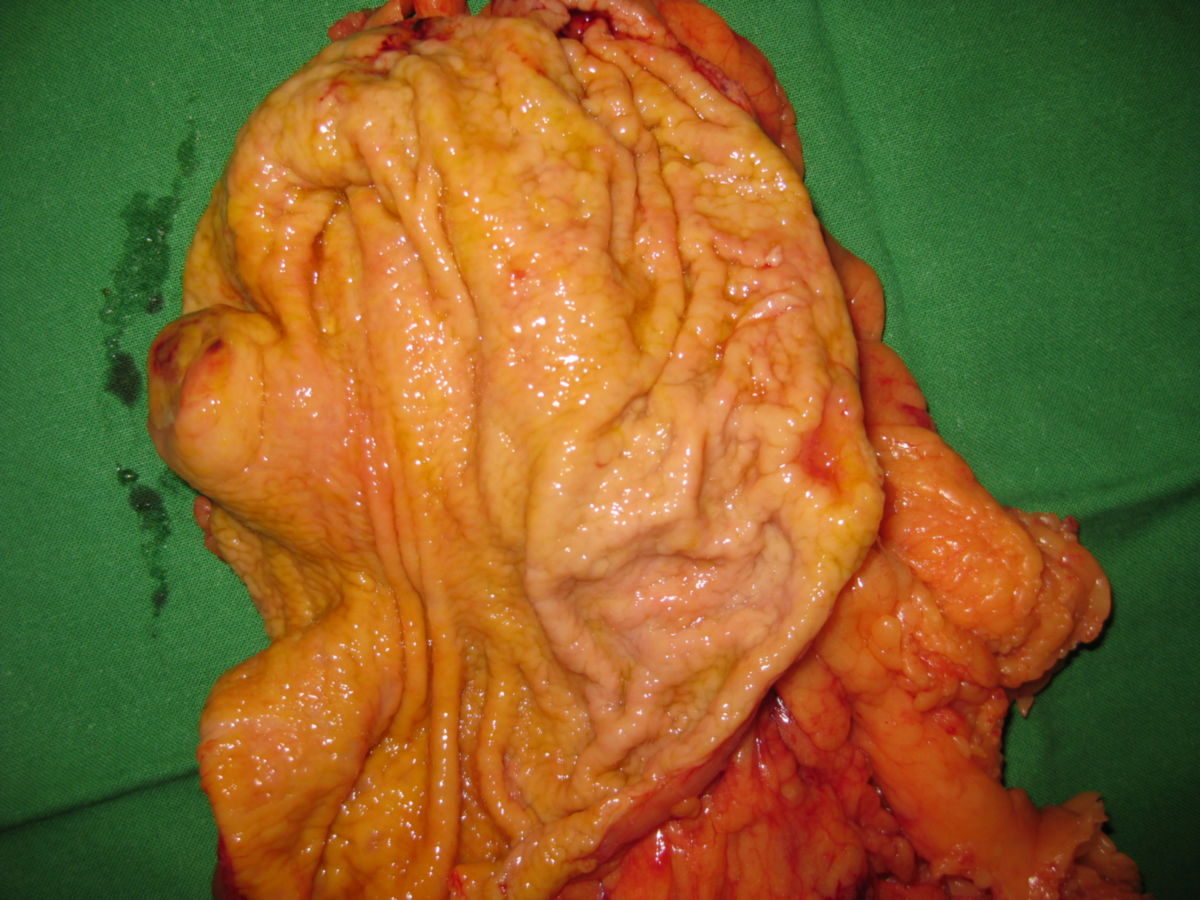
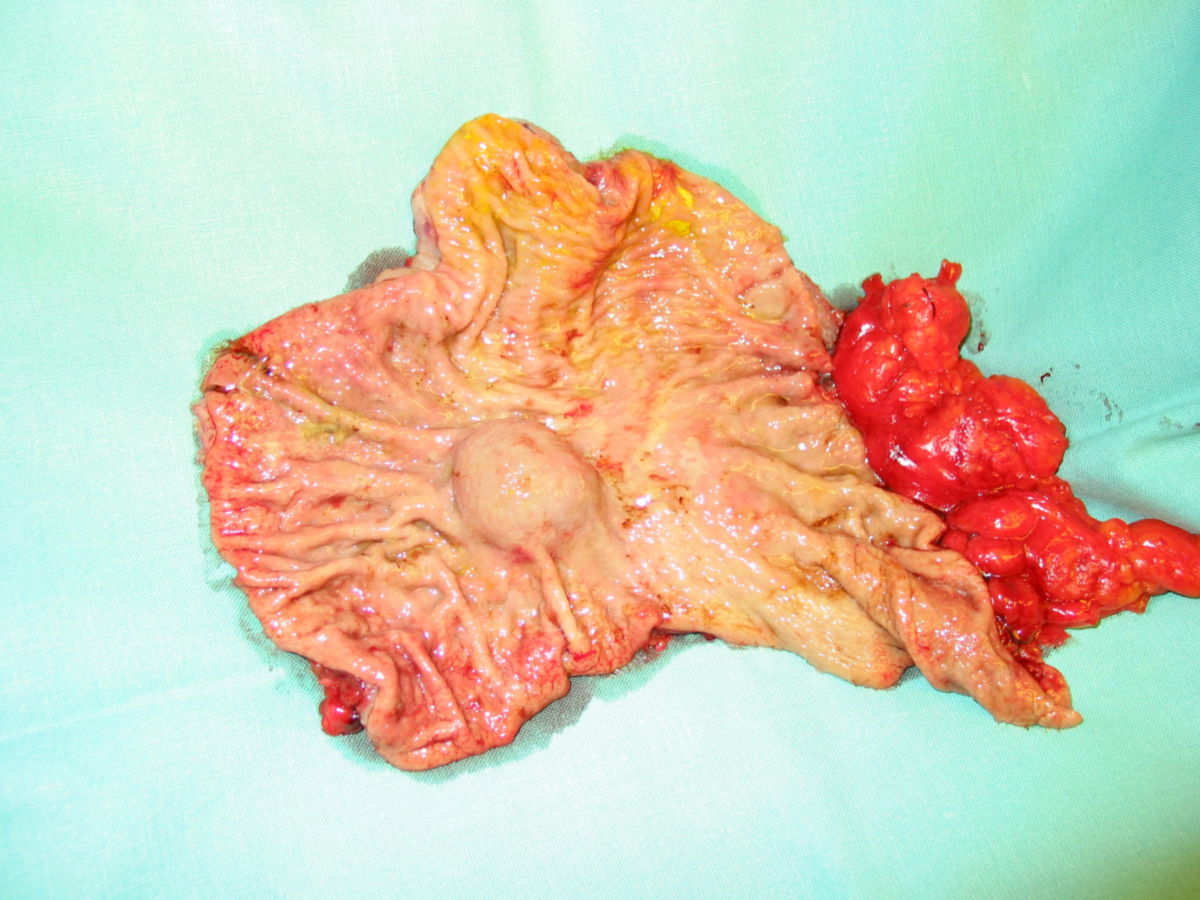
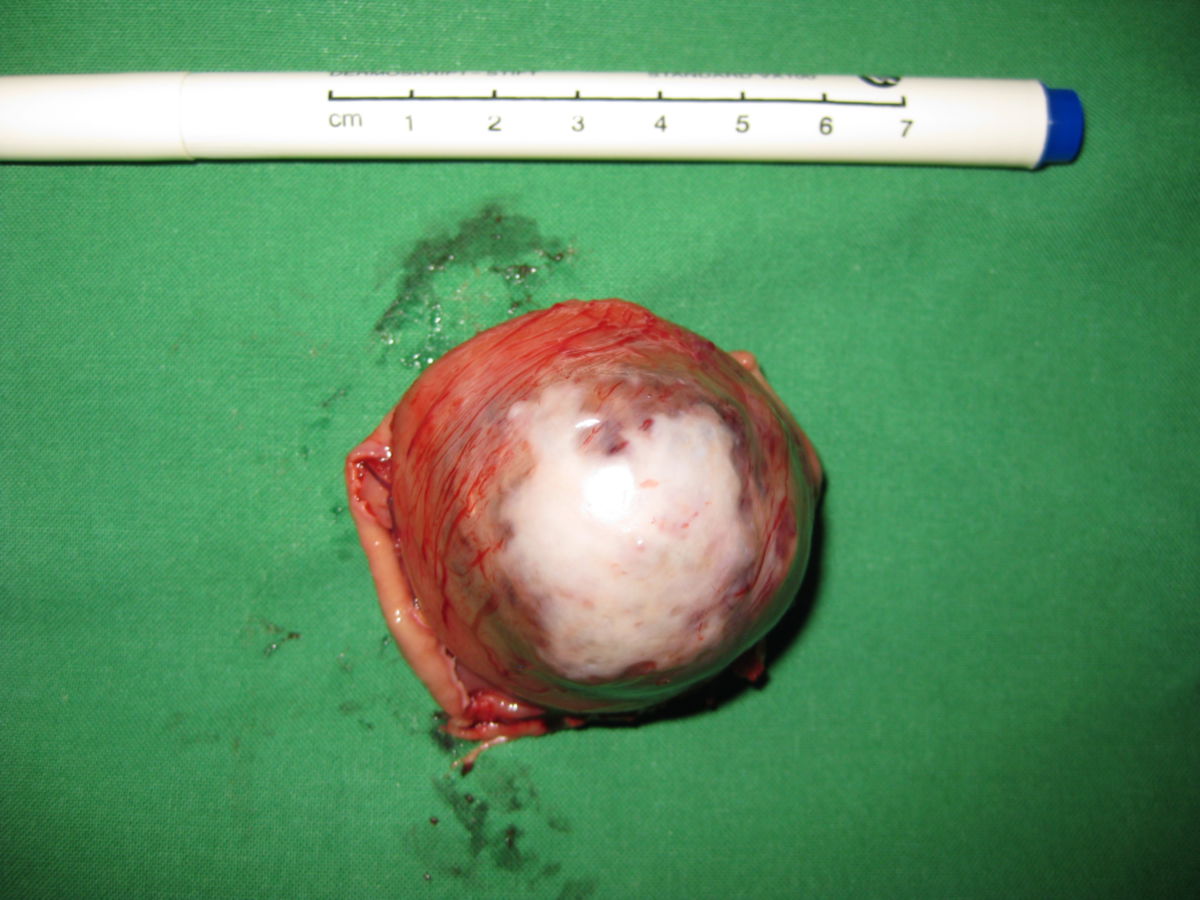
Die Entdeckung, dass man GIST anhand des so genannten KIT-Proteins nachweisen kann (Hirota 1998), half festzustellen, dass sich GIST aus den so genannten "Cajal-Zellen" oder gemeinsamen Vorstufen entwickeln. Dies sind "Schrittmacher-Zellen", die in der äußeren Wand des Verdauungstraktes angeordnet sind. GIST entstehen also in der Wand der Verdauungsorgane. Sie wachsen von dort meist nicht in die Organe hinein, sondern breiten sich ungehindert in den Bauchraum aus. Daher werden sie oft auch erst sehr spät und teilweise in enormer Größe diagnostiziert.
Etwa die Hälfte der Patienten mit neu diagnostiziertem GIST weisen bereits Metastasen auf. GIST-Metastasen findet man am häufigsten in der Leber (ca. 65 %) oder im Peritoneum (ca. 20 %) – sehr selten in anderen Organen. Grundsätzlich sollte das Wort "benigne" in Verbindung mit GIST nicht gebraucht werden, da alle GIST potenziell maligne sind, d.h. alle GIST – auch kleine Tumoren – durchaus zu Streuherden führen können.
Epidemiologie
Das mediane Erkrankungsalter liegt bei etwa 65–70 Jahren. Die Inzidenz beträgt ca. 10–15 Neuerkrankungen pro 1 Million Einwohner und Jahr. In Deutschland entspricht dies etwa 1.500–2.000 Neuerkrankungen jährlich. Männer und Frauen sind etwa gleich häufig betroffen. Vereinzelt tritt die Erkrankung bereits im Kindes- oder Jugendalter auf (pädiatrische GIST).
Pathogenese
Der Entstehungsgrund für GIST ist bisher (2026) nicht geklärt. Entscheidend für die Pathogenese scheint eine Mutation im KIT- oder PDGF-Rezeptor alpha zu sein, die zu einer kontinuierlichen ligandenunabhängigen Aktivität der Rezeptor-Tyrosinkinase führt. Am häufigsten betroffen sind KIT Exon 11 (ca. 70 %), KIT Exon 9 (ca. 10 %) und PDGFR alpha Exon 18 (ca. 6 %). Neben KIT- und PDGFRA-Mutationen existieren weitere molekulare Subgruppen (z.B. SDH-defiziente, NF1-assoziierte oder BRAF-mutierte GIST), die unter dem Begriff "Wildtyp-GIST" zusammengefasst werden, jedoch biologisch heterogen sind.
Die Ermittlung des Mutationsstatus im Rahmen der Diagnostik ist wichtig, da die Mutation auf das Therapieansprechen und die Prognose einen entscheidenden Einfluss hat.
Aufgrund der therapeutischen Konsequenzen sollte eine Mutationsanalyse von einem erfahrenen Pathologen durchgeführt werden.
Lokalisation
Die wesentlichen Lokalisationen sind:
- 50–60 % im Magen
- 20–30 % im Dünndarm
- 5–12 % im Dickdarm und Rektum
- ≤ 1 % in der Speiseröhre
Symptome
Die ersten Anzeichen bei Patienten mit GIST hängen von der Tumorgröße und der Lokalisation des Primärtumors ab. Ein signifikanter Anteil kleiner und asymptomatischer Tumoren wird zufällig bei diagnostischen Maßnahmen (Endoskopie) oder im Rahmen anderer operativer Eingriffe entdeckt. Ein relevanter Anteil wird im Rahmen einer Notfalloperation aufgrund von gastrointestinaler Obstruktion oder Tumorperforation mit abdomineller Blutung diagnostiziert.
Die häufigsten Symptome sind:
- Tumoren im Magen oder Duodenum
- Schmerzen (50–70 % der Betroffenen)
- Gastrointestinale Blutungen (20–50 %)
- Völlegefühl
- Dünndarm-Tumoren
- Schmerzen
- Blutung
- Obstruktion
- Dickdarm-Tumoren
- Blutauflagerung Stuhl
- Obstruktion
- Ösophagus
- Unspezifische Allgemeinsymptome
Diagnostik
Die Diagnose basiert auf der Kombination aus klinischem Befund, bildgebender Diagnostik, Histologie, Immunhistochemie und Molekulargenetik.
Die bildgebende Diagnostik dient der Erfassung des Primärtumors sowie dem Staging. Die Methode der Wahl ist die kontrastmittelverstärkte Computertomographie (CT) von Thorax, Abdomen und Becken. Sie ermöglicht die Beurteilung der lokalen Tumorausdehnung sowie den Nachweis von Metastasen, insbesondere in Leber und Peritoneum. In ausgewählten Fällen kann eine PET-CT zur Beurteilung des Therapieansprechens sinnvoll sein.
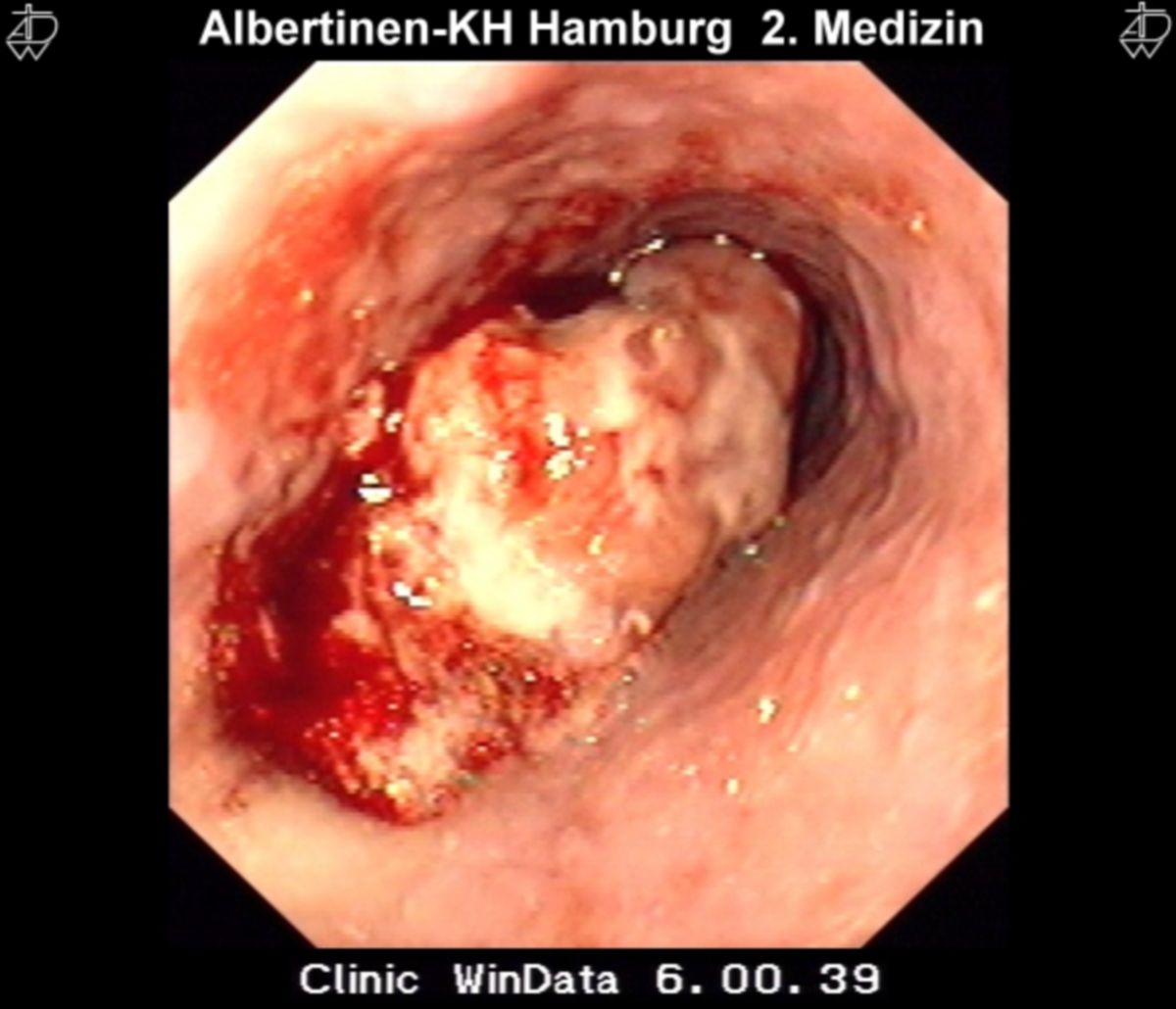
Je nach Lokalisation des Primärtumors kommen endoskopische und endosonographische Verfahren zum Einsatz. Diese ermöglichen die Gewinnung von Biopsien zur histologischen Sicherung, sofern dies technisch möglich ist und keine relevante Gefahr einer Tumordissemination besteht.
Die histopathologische Diagnose erfolgt anhand typischer morphologischer Merkmale (spindelzellig, epitheloid oder gemischtzellig) sowie immunhistochemischer Marker. Charakteristisch ist die Expression von KIT (CD117) und DOG1, die bei der Mehrzahl der GIST nachweisbar sind.
Die molekulargenetische Untersuchung mit Bestimmung des KIT- bzw. PDGFRA-Mutationsstatus ist bei allen Patienten, bei denen eine systemische Therapie in Betracht kommt, obligater Bestandteil der Initialdiagnostik.
Im Wesentlichen unterscheidet man folgende klinische Situationen:
- Lokal begrenzter, operabler Primärtumor (keine Metastasierung)
- Lokal fortgeschrittener bzw. primär nicht resektabler Primärtumor
- Metastasierter bzw. inoperabler GIST
- Progression der Erkrankung unter systemischer Therapie
Diagnostik, Therapie und Verlaufskontrolle sollten interdisziplinär erfolgen. Aufgrund der Seltenheit der Erkrankung und der Komplexität der Therapie ist die Behandlung in einem spezialisierten Sarkomzentrum empfohlen.
Therapie
Die Therapie des GIST erfolgt multimodal und orientiert sich an Tumorstadium, Resektabilität, Mutationsstatus sowie dem individuellen Rezidivrisiko. Ziel ist sowohl die lokale Tumorkontrolle als auch die Vermeidung bzw. Behandlung von Metastasen.
Beobachtendes Vorgehen
Bei kleinen, asymptomatischen GIST des Magens mit einer Größe unter 2 cm kann in ausgewählten Fällen ein abwartendes Vorgehen ("watch and wait") mit regelmäßigen endoskopischen und endosonographischen Kontrollen erwogen werden. Voraussetzung ist ein fehlendes Tumorwachstum. Für GIST anderer Lokalisationen wird aufgrund eines höheren Progressionsrisikos in der Regel eine primäre Resektion empfohlen.
Chirurgische Therapie
Die chirurgische Resektion ist die Therapie der Wahl bei lokal begrenzten, resektablen Tumoren. Angestrebt wird eine vollständige (R0-Resektion) ohne Tumorruptur. Eine systematische Lymphadenektomie ist aufgrund der seltenen lymphogenen Metastasierung in der Regel nicht erforderlich.
Bei lokal fortgeschrittenen Tumoren kann eine primäre Operation technisch schwierig oder mit erheblicher Morbidität verbunden sein. In diesen Fällen kann eine präoperative (neoadjuvante) Therapie zur Tumorverkleinerung erfolgen, um die Resektabilität zu verbessern.
Auch bei metastasierter Erkrankung kann in ausgewählten Fällen eine chirurgische Entfernung von Metastasen (Metastasektomie) erwogen werden, insbesondere bei gutem Ansprechen auf eine systemische Therapie. Der Nutzen ist jedoch nicht eindeutig durch prospektive Studien belegt.
Medikamentöse Therapie
Die systemische Therapie mit Tyrosinkinaseinhibitoren (TKI) ist ein zentraler Bestandteil der Behandlung von GIST und wird sowohl in der adjuvanten als auch in der palliativen Situation eingesetzt.
In der neoadjuvanten Situation kommt Imatinib zum Einsatz, wenn eine primäre Resektion nicht möglich oder mit erheblicher Funktionseinschränkung verbunden wäre. Ziel ist die Tumorverkleinerung zur Ermöglichung einer operativen Therapie. Voraussetzung ist eine Imatinib-sensitive Mutation. Bei Vorliegen einer PDGFRA-D842V-Mutation kann alternativ Avapritinib eingesetzt werden.
Nach vollständiger Resektion wird bei Patienten mit erhöhtem Rezidivrisiko eine adjuvante Therapie mit Imatinib empfohlen. Die Standarddauer beträgt drei Jahre. Patienten mit niedrigem Risiko profitieren in der Regel nicht von einer adjuvanten Therapie.
Bei metastasierten oder inoperablen GIST ist die medikamentöse Therapie die Behandlung der ersten Wahl. Die Therapie erfolgt in einer sequenziellen Abfolge:
- Imatinib (Erstlinientherapie)
- Sunitinib (Zweitlinientherapie)
- Regorafenib (Drittlinientherapie)
- Ripretinib (Viertlinientherapie)
Bei einer PDGFRA-D842V-Mutation gilt auch hier Avapritinib als Therapie der Wahl. Die Behandlung wird in der Regel kontinuierlich bis zum Nachweis einer Progression fortgeführt, da Therapiepausen mit einem hohen Risiko für ein rasches Tumorwachstum verbunden sind.
Bei Progress unter Imatinib kann zunächst eine Dosiserhöhung erwogen werden. Bei weiterer Progression erfolgt der Wechsel auf die nächste Therapielinie. Zusätzlich können bei lokalisierter Progression ergänzend lokale Therapieverfahren eingesetzt werden.
Strahlentherapie
Die Strahlentherapie hat bei GIST keinen etablierten Stellenwert. Sie kann in Einzelfällen im palliativen Setting, beispielsweise bei Knochenmetastasen oder lokal symptomatischen Tumoren, eingesetzt werden.
Lokoregionale Verfahren
Bei selektierten Patienten, insbesondere bei oligometastatischer oder oligoprogressiver Erkrankung, können ergänzend lokoregionäre Therapieverfahren zum Einsatz kommen. Hierzu zählen unter anderem:
- chirurgische Resektion einzelner Läsionen
- Radiofrequenzablation (RFA)
- transarterielle Verfahren (z. B. TAE, SIRT)
Nachsorge
Nach abgeschlossener Therapie ist eine strukturierte Nachsorge erforderlich, da Rezidive insbesondere in den ersten Jahren nach Primärbehandlung auftreten können. Die Verlaufskontrolle dient der frühzeitigen Detektion von Lokalrezidiven oder Metastasen sowie der Überwachung des Therapieansprechens unter systemischer Behandlung.
Die Nachsorge erfolgt in der Regel mittels kontrastmittelverstärkter Computertomographie von Abdomen und Becken. Die Untersuchungsintervalle richten sich nach dem individuellen Rezidivrisiko:
- hohes Risiko: alle 3–6 Monate in den ersten 3–5 Jahren
- niedriges Risiko: längere Intervalle möglich
- bei metastasierter Erkrankung erfolgen Verlaufskontrollen meist alle 3–4 Monate.
Ein wesentlicher Bestandteil der Nachsorge ist zudem das Management der systemischen Therapie. Unter Behandlung mit Tyrosinkinaseinhibitoren ist eine gute Compliance entscheidend für den Therapieerfolg. Dies erfordert eine strukturierte Patientenaufklärung sowie ein konsequentes Nebenwirkungsmanagement, um Therapieabbrüche zu vermeiden und die Wirksamkeit langfristig zu erhalten.
Prognose
Patienten mit lokal begrenzter Erkrankung und vollständiger R0-Resektion haben eine vergleichsweise gute Prognose. Das Rezidivrisiko steigt jedoch mit zunehmender Tumorgröße und hoher Mitoserate deutlich an.
Unter moderner systemischer Therapie mit Tyrosinkinaseinhibitoren hat sich die Prognose insbesondere bei metastasierter Erkrankung erheblich verbessert. Die mediane Überlebenszeit liegt mittlerweile bei mehreren Jahren.
Bestimmte molekulare Subtypen beeinflussen die Prognose zusätzlich: So sind beispielsweise PDGFRA-D842V-mutierte Tumoren gegenüber Imatinib resistent, weisen jedoch häufig einen eher indolenten Verlauf auf, während andere Mutationen mit aggressiverem Krankheitsverlauf assoziiert sein können.