Hutchinson-Gilford-Syndrom







Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
Loslegennach den britischen Chirurgen Jonathan Hutchinson (1828–1913) und Hastings Gilford (1861–1941)
Synonyme: Progeria infantilis, Hutchinson-Progerie, Gilford-Syndrom, greisenhafter Zwergwuchs, Progerie 1, Vergreisungssyndrom
Englisch: infantile progeria, Hutchinson-Gilford progeria syndrome (HGPS)
Definition
Das Hutchinson-Gilford-Syndrom, kurz HGPS, ist eine seltene, autosomal-dominant vererbte Erkrankung, die zu einem massiven und sehr früh einsetzenden Alterungsprozess (Progerie) von Haut, Skelett und Blutgefäßen in der frühen Kindheit führt. Die ursächliche Mutation im LMNA-Gen entsteht nahezu immer de novo.
Epidemiologie
Das Hutchinson-Gilford-Syndrom ist eine sehr seltene Erkrankung. Die weltweite Prävalenz wird auf etwa 1 : 20.000.000 geschätzt.[1] Da die verursachende Mutation nahezu immer neu entsteht, ist das Wiederholungsrisiko für Geschwister gering. Ein überdurchschnittlich hohes Alter des Vaters gilt als Risikofaktor (Paternal age effect).
Ätiologie
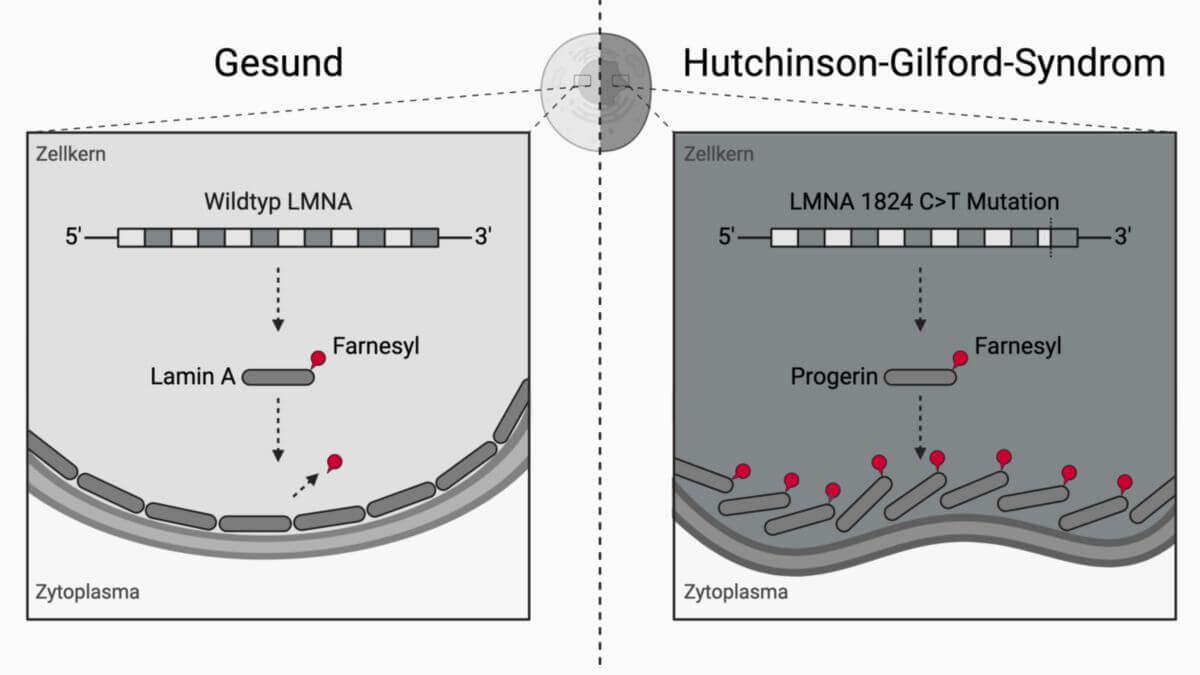
Dem Hutchinson-Gilford-Syndrom liegt ein Defekt des LMNA-Gens auf Chromosom 1 (1q22) zugrunde, das für das Protein Lamin A/C kodiert. Lamin A/C ist ein wesentlicher Bestandteil der Kernlamina an der Innenseite der Kernhülle, der als Anknüpfungspunkt des Filamentsystems und als Regulator der Gentranskription fungiert.
Bei etwa 90 % der Patienten liegt eine de novo entstandene stille Punktmutation im Codon 608 (c.1824C>T, p.G608G) vor, durch die eine kryptische Splicestelle aktiviert wird. Dabei gehen 150 Nukleotide des Exons 11 verloren, was zum Verlust von 50 internen Aminosäuren im späteren Protein führt. Dieses verkürzte, dauerhaft farnesylierte Protein wird Progerin genannt und reichert sich in der Kernmembran an, wo es die Kernarchitektur destabilisiert.[2]
Neben dem Hutchinson-Gilford-Syndrom können Mutationen im LMNA-Gen auch andere Erkrankungen (Laminopathien) auslösen. Der Grund für die Unterschiede der jeweiligen Krankheitsbilder ist bisher (2026) noch nicht abschließend verstanden.
Symptome
Patienten mit Hutchinson-Gilford-Syndrom weisen bereits im ersten Lebensjahr Wachstumsdefizite auf. Das Vollbild der Erkrankung manifestiert sich meist um das dritte Lebensjahr.
Die Patienten bleiben kleinwüchsig und weisen Skelettdeformitäten auf. Typische muskuloskelettale Befunde sind Gelenkkontrakturen, Coxa valga, Akroosteolyse und eine Resorption der distalen Klavikula. Hinzu kommen eine relative Makrozephalie, prominente Kopfhautvenen und eine schmale, vorspringende, schnabelförmige Nase.
Die Haut erscheint durchscheinend, das Unterhautfettgewebe fehlt fast vollständig (Lipodystrophie). Die Hautanhangsgebilde sind in Form von Alopezie und Nageldystrophie betroffen. Zahnmedizinisch kommt es zu einem verzögerten Durchbruch und verzögerten Verlust der Milchzähne. Der Durchbruch der bleibenden Zähne ist unvollständig oder bleibt aus.
Die Sinnesorgane sind in Form von Schwerhörigkeit und Lagophthalmus beeinträchtigt.
Eine Geschlechtsentwicklung durch Pubertät ist gestört. Die sekundären Geschlechtsmerkmale entwickeln sich meist unvollständig, die Patienten sind infertil. Die Intelligenz entwickelt sich in aller Regel altersgemäß. Klinisch führend ist eine früh einsetzende, generalisierte Arteriosklerose.
Diagnostik
Die Verdachtsdiagnose wird anhand des charakteristischen klinischen Bildes gestellt (Wachstumsstörung, Alopezie, Lipodystrophie, typische progeroide Fazies). Die Diagnose wird durch den molekulargenetischen Nachweis einer heterozygoten, zur Progerinbildung führenden pathogenen LMNA-Variante (z.B. c.1824C>T) gesichert.
Therapie
Eine kausale Therapie ist bislang (2026) nicht verfügbar. Die Behandlung erfolgt überwiegend symptomatisch und richtet sich auf die Prophylaxe und Therapie der kardiovaskulären Komplikationen. Die umfasst u.a. Physio- bzw. Ergotherapie sowie eine adäquate Zahn-, Augen- und Hörversorgung.
Mit dem Farnesyltransferase-Hemmer Lonafarnib steht seit 2020 (FDA) bzw. 2022 (EMA) erstmals eine krankheitsmodifizierende Therapie zur Verfügung. Lonafarnib vermindert durch Hemmung der Farnesyltransferase eine Anreicherung von farnesyliertem Progerin und damit seine toxischen Wirkungen. In der Zulassungsdatenlage führte die Behandlung zu einer signifikanten Verlängerung der Überlebenszeit.[3][1] Die Zulassung besteht für Patienten ab einem Alter von einem Jahr.
Prognose
Die Prognose ist ungünstig. Unbehandelt versterben die Patienten im Mittel um das 14. bis 15. Lebensjahr, meist an den Folgen der akzelerierten Arteriosklerose – vor allem durch Myokardinfarkt oder Schlaganfall.[3] Unter Therapie mit Lonafarnib verlängert sich die Lebenserwartung um durchschnittlich 3-4 Jahre.
Weblink
Quellen
- ↑ 1,0 1,1 Dhillon S. Lonafarnib: First Approval. Drugs. 2021;81(2):283–289.
- ↑ Eriksson M et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423(6937):293–298.
- ↑ 3,0 3,1 Gordon LB et al. Association of Lonafarnib Treatment vs No Treatment With Mortality Rate in Patients With Hutchinson-Gilford Progeria Syndrome. JAMA. 2018;319(16):1687–1695.