Akute intermittierende Porphyrie






Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenSynonym: AIP
Englisch: acute intermittent porphyria
Definition
Unter der akuten intermittierenden Porphyrie, kurz AIP, versteht man eine autosomal-dominant vererbte akute hepatische Porphyrieform, die zu Symptomen im Gastrointestinaltrakt, im ZNS und im Herz-Kreislauf-System führt.
Epidemiologie
Die Prävalenz der akuten intermittierenden Porphyrie beträgt 5 bis 10 pro 100.000 Einwohner. Frauen sind aufgrund hormoneller Faktoren dreimal so häufig betroffen wie Männer, wobei der Erkrankungsgipfel im dritten Lebensjahrzehnt liegt.
Die Erkrankung tritt weltweit auf, es gibt jedoch eine Häufung in Skandinavien und Großbritannien.
Ätiopathogenese
Die Erkrankung beruht auf einem autosomal-dominant vererbten Defekt der Hydroxymethylbilan-Synthase, einem zytosolischen Enzym der Hämsynthese. Dies führt zu einer Reduktion der Enzymfunktion um > 50 %, was unter normalen Umständen jedoch noch für die Hämsynthese ausreichend ist.
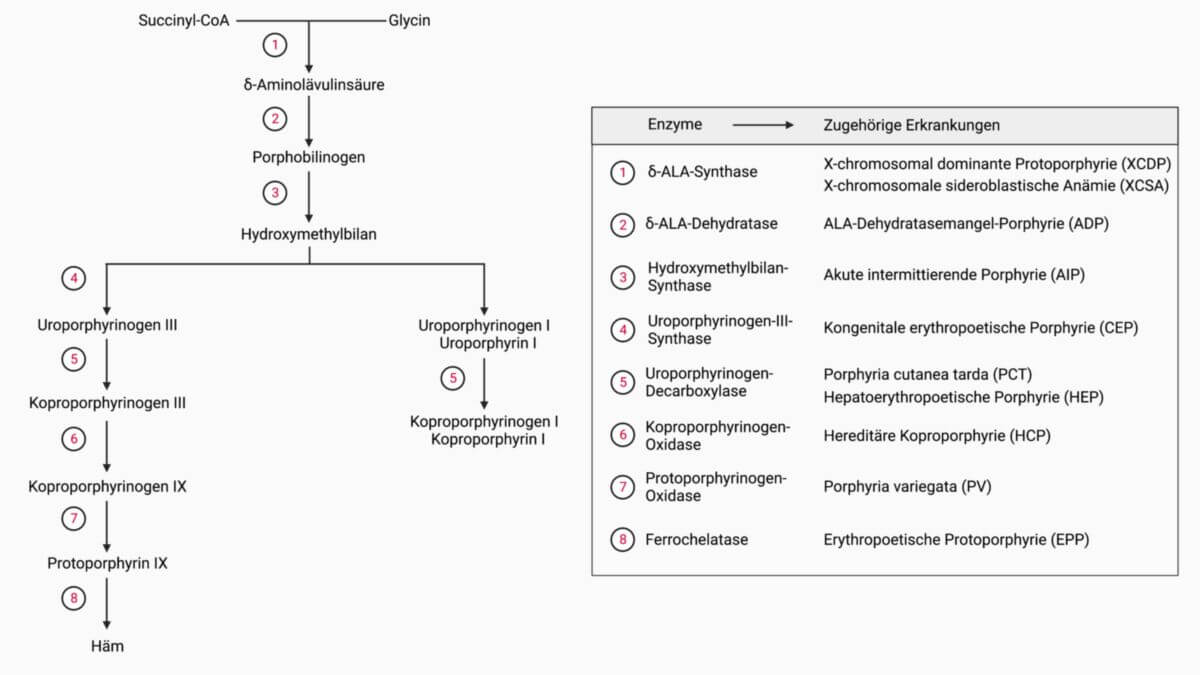
Steigt der Häm-Bedarf des Körpers, beispielsweise durch eine Induktion von Cytochrom-P450-Enzymen, kommt es zur Mehrexpression des vorangehenden Enzyms δ-Aminolävulinat-Synthase 1 (δ-ALAS 1, Schrittmacherenzym der hepatischen Hämsynthese). Die Restaktivität der Hydroxymethylbilan-Synthase reicht dann nicht mehr aus, um die anfallenden Metaboliten zu verstoffwechseln. δ-Aminolävulinsäure (δ-ALA) und Porphobilinogen (PBG) akkummulieren. Sie verteilen sich systemisch und werden letztlich renal exkretiert. Weswegen diese Stoffe zu untenstehender Symptomatik führen, ist unklar. Für δ-ALA wird eine Interferenz mit dem strukturähnlichen Neurotransmitter GABA angenommen[1], jedoch finden sich auch histologische Schäden im Nervengewebe (Axonschäden und Demyelinisierung)[2].
Klinik
Die Erkrankung besitzt eine unvollständige Penetranz. Nur ca. 20 % der Mutationsträger werden zeitlebens symptomatisch. Bei diesen Patienten kommt es zu anfallsartigen Krankheitsattacken, die Tage bis Wochen anhalten. Ein solcher Ausbruch der Erkrankung kann durch Infektionen, Alkohol, Operationen, Medikamente, prämenstruelle Hormonveränderungen, Stress und Hypoglykämien ausgelöst werden. Meist zeigt sich die erste Attacke in oder nach der Pubertät.
Im akuten Anfall sind folgende Symptome möglich[2]:
- Abdominalsymptome
- kolikartige Bauchschmerzen bei weicher Bauchdecke (oft Initialsymptom)
- Obstipation bis hin zum Ileus
- Übelkeit und Erbrechen
- Hypertonie und Tachykardie
- neurologische Symptome
- hyperaktives Delir, Agitation, Halluzinationen, Bewusstseinsstörungen bis Koma
- epileptische Anfälle
- periphere sensorische Neuropathie mit Rücken-/Extremitätenschmerzen und Parästhesien
- motorische Neuropathie mit Paresen, oft an den Armen beginnend
- SIADH mit Hyponatriämie
Auf lange Sicht besteht ein erhöhtes Risiko für eine arterielle Hypertonie, eine chronische Niereninsuffizienz und ein hepatozelluläres Karzinom.
Im Gegensatz zu anderen Porphyrieformen findet sich keine Photodermatose.
Diagnostik
Die Familienanamnese ist aufgrund der geringen Penetranz und der Möglichkeit von Neumutationen oft unauffällig.
Der Urin der betroffenen Patienten kann sich bei längerem Luftkontakt rötlich anfärben. Als Screeningmaßnahme wird der Urin im Anfall auf Porphobilinogen untersucht (Hoesch-Test). Anschließend erfolgt die quantitative Bestimmung von Porphobilinogen und δ-Aminolävulinsäure im Urin, in Relation zum Kreatinin-Quotienten (PBG oder δ-ALA in µg/g Kreatinin). Eine Erhöhung auf mindestens das vierfache der Norm macht eine Porphyrie wahrscheinlich. Bei geringeren Werten mit passender Symptomatik sollten alternative Differenzialdiagnosen in Betracht gezogen werden. Normwertiges PBG und δ-ALA schließen eine akute intermittierende Porphyrie als Ursache der Beschwerden aus.
Die Spiegel dienen nicht nur der Diagnosestellung, sondern können auch zur Verlaufskontrolle herangezogen werden.
Zum Nachweis einer Mutationsträgerschaft (z.B. bei Familienangehörigen oder in symptomfreien Intervallen) kann außerdem die erythrozytäre Hydroxymethylbilan-Synthase-Aktivität bestimmt werden oder eine Zielgensequenzierung erfolgen.
Differenzialdiagnose
Differenzialdiagnostisch sollte an eine Panarteriitis nodosa, neurologische Erkrankungen, an eine Bleivergiftung sowie andere Ursachen des akuten Abdomens gedacht werden. Auch andere akute hepatische Porphyrien sind zu bedenken, selbst wenn diese deutlich seltener auftreten.
Therapie
Alkoholkarenz und das Absetzen porphyrinogener Medikamente sind zwingend erforderlich.
Das akute Auftreten der Erkrankung wird mit Glukoseinfusionen und forcierter Diurese sowie in schweren Fällen mit Häm-Arginin behandelt. Bei Schmerzen können zusätzlich ASS oder Morphinderivate gegeben werden. Eine Hypertonie wird mit Beta-Blockern behandelt, Unruhe und Brechreiz mit einem niederpotenten Neuroleptikum.
In der Latenzphase kann bei deutlicher Erhöhung der Urinmetabolite eine Hämarginin-Intervalltherapie erfolgen. Sinnvoll ist auch das Ausstellen eines Patientenausweises mit möglichen und kontraindizierten Medikamenten.
Seit 2019 steht mit Givosiran eine RNA-Interferenz-Therapie zur Verfügung, die die hepatische ALAS1-Expression senkt und die Attackenfrequenz deutlich reduzieren kann.
Prognose
Bei frühzeitiger Diagnose und konsequenter Triggervermeidung ist die Prognose günstig. Unbehandelte schwere Attacken können jedoch zu irreversiblen neurologischen Schäden oder lebensbedrohlichen Komplikationen führen.
Quellen
Literatur
- Suttorp et al., Harrisons Innere Medizin, Thieme Verlag, 20. Auflage, 2020