Leberzirrhose











von altgriechisch: κιρρός ("kirrós") - gelb-orange, gelblich
Synonym: Schrumpfleber
Englisch: cirrhosis, liver cirrhosis
Definition
Die Leberzirrhose ist das Endstadium chronischer Lebererkrankungen. Sie ist durch einen diffusen, fibro-nodulären Umbau der Leber mit Verlust der normalen Läppchen- und Gefäßarchitektur gekennzeichnet. Funktionelle Folgen sind Leberinsuffizienz, portale Hypertension und die Ausbildung intrahepatischer porto-systemischer Shunts.
Hintergrund
Die Leberzirrhose stellt den gemeinsamen Endpunkt verschiedener chronischer Lebererkrankungen dar. Sie entsteht als Folge einer anhaltenden, meist entzündlich bedingten Schädigung, die eine kontinuierliche Regeneration und gleichzeitig fortschreitende Fibrosierung induziert. Dieser Prozess führt zu einem diffusen Umbau der Leberarchitektur mit Verlust der physiologischen Läppchenstruktur und Störung der intrahepatischen Gefäßorganisation.
Klinisch wird die Leberzirrhose heute im Konzept der Advanced Chronic Liver Disease (ACLD) betrachtet. Dabei werden eine kompensierte Phase – in der die Leberfunktion noch weitgehend erhalten ist – und eine dekompensierte Phase des Akut-auf-chronischen Leberversagens (ACLF) unterschieden.
Die Fibroseentwicklung ist dynamisch: Bei erfolgreicher Beseitigung der auslösenden Noxe kann es in frühen Stadien zu einer teilweisen Rückbildung der Fibrose kommen. Im weit fortgeschrittenen Stadium persistiert jedoch in der Regel eine irreversibel gestörte Architektur, auch wenn das Risiko für Komplikationen abnimmt.
Epidemiologie
Die Leberzirrhose ist weltweit ein bedeutendes Gesundheitsproblem. Die globale Prävalenz wird auf etwa 1 % der Bevölkerung geschätzt, mit deutlichen regionalen Unterschieden in Abhängigkeit von den zugrunde liegenden Ätiologien (siehe dort). Im Jahr 2019 wurden weltweit rund 2,05 Millionen Neuerkrankungen registriert. Die Inzidenz in westlichen Industrieländern (Europa und USA) beträgt etwa 250 Fälle pro 100.000 Einwohner pro Jahr.
Die Leberzirrhose gehört zu den führenden Ursachen leberbedingter Mortalität. Sie ist für rund 2,4 % aller Todesfälle weltweit verantwortlich. Die 10-Jahres-Mortalität der Leberzirrhose liegt etwa zwischen 30 und 60 %.[1] In westlichen Ländern zählt sie zu den häufigsten Indikationen für eine Lebertransplantation. Das Erkrankungsrisiko steigt deutlich mit zunehmendem Alter. Männer sind etwa eineinhalbmal häufiger betroffen als Frauen.
Einteilung
Die Einteilung der Leberzirrhose hat das Ziel, den Schweregrad der Erkrankung, die Prognose und die therapeutische Strategie festzulegen. Sie berücksichtigt sowohl morphologische Veränderungen, klinisch-funktionelle Aspekte als auch hämodynamische Parameter.
...nach klinischen Stadien
Nach dem Konzept der Advanced Chronic Liver Disease (ACLD) wird die Erkrankung anhand des klinischen Verlaufs in folgende Stadien unterteilt:
- Kompensierte Zirrhose: Leberfunktion ausreichend, keine klinisch relevanten Komplikationen. Patienten sind oft asymptomatisch oder haben unspezifische Symptome.
- Dekompensierte Zirrhose: Auftreten mindestens einer typischen Komplikation wie Aszites, Varizenblutung, hepatische Enzephalopathie oder Ikterus. Medianes Überleben reduziert auf 1–2 Jahre ohne Transplantation.
Die Übergangsphase zur Dekompensation wird maßgeblich durch die Entwicklung einer klinisch signifikanten portalen Hypertension (CSPH) definiert, die in der Regel einem Lebervenendruckgradient (HVPG) ≥ 10 mmHg entspricht. Bei HVPG ≥ 12 mmHg steigt das Risiko für Varizenblutungen deutlich an.
...nach Child-Pugh-Kriterien
Die Leberzirrhose kann nach ihrem Schweregrad anhand der Child-Pugh-Kriterien in drei Stadien (Child A bis C) differenziert werden. Berücksichtigt werden fünf Parameter (Bilirubin, Albumin, INR/Quick, Aszites, hepatische Enzephalopathie):
- Child A (5–6 Punkte): gute Leberfunktion, 1-Jahres-Überleben > 90 %
- Child B (7–9 Punkte): mäßig eingeschränkte Funktion
- Child C (10–15 Punkte): stark eingeschränkte Funktion, 1-Jahres-Überleben < 50 %
Der Child-Pugh-Score wird häufig genutzt, um das Operationsrisiko oder die Indikation für bestimmte Therapien (z.B. TIPS) einzuschätzen.
...nach MELD-Score
Der MELD-Score ist das zentrale System zur Priorisierung von Patienten auf der Lebertransplantationsliste. Er basiert auf drei Laborwerten (Serumkreatinin, Gesamtbilirubin, INR) Ergänzend kann bei Patienten mit Aszites der Serumnatriumwert (MELD-Na) einbezogen werden, um die Prognosegenauigkeit zu verbessern. Ein höherer MELD-Score korreliert mit einer höheren 3-Monats-Mortalität (z.B. MELD 10 geht mit 3-Monats-Mortalität von ca. 6 % einher).
...nach Baveno-Kriterien
Die Baveno-Kriterien orientieren sich am Vorhandensein einer klinisch signifikanten portalen Hypertension (CSPH), die wichtig für Screening-Strategien und therapeutische Entscheidungen sind:
- keine CSPH: HVPG < 10 mmHg, keine Varizen, sehr geringes Blutungsrisiko
- CSPH: HVPG ≥ 10 mmHg, Risiko für Varizen und Dekompensation steigt
- Klinisch relevante CSPH: Nachweisbare Varizen, Splenomegalie, Thrombozytopenie
- Hochgradige CSPH: HVPG ≥ 12 mmHg, hohes Risiko für Varizenblutung
...nach CLIF-C ACLF-Score
Das Akut-auf-chronische Leberversagen (ACLF) beschreibt eine akute Verschlechterung der Leberfunktion bei bestehender Zirrhose, häufig ausgelöst durch Infektionen, Alkohol, Blutungen oder Medikamente. Der CLIF-C-ACLF-Score bewertet Leberfunktion, Nierenfunktion, Gerinnung, Zirkulation, Atmung und Gehirnfunktion. Je nach Anzahl und Schweregrad der Organversagen erfolgt die Einteilung in ACLF-Grad 1 bis 3. CLIF-C ACLF ≥ 64 gilt als Indikator für eine sehr schlechte Prognose.
...nach Pathohistologie
Das pathohistologische Bild ist unabhängig von der Ätiologie. Es kommt zur Ausbildung von portoportalen und portozentralen Bindegewebssepten und Regeneratknoten.
Die histologische Einteilung ist wie folgt:
| Bezeichnung | Durchmesser der Regeneratknötchen | Hintergrund |
|---|---|---|
| Mikronoduläre Leberzirrhose | bis 3 mm | typisch bei alkoholbedingter Leberzirrhose |
| Makronoduläre Leberzirrhose | 3 mm bis 3 cm | häufig bei posthepatitischen Formen |
| Gemischtknotige Leberzirrhose | variabel | häufig im Spätstadium |
...nach Ishak-Score
Der Ishak-Score wird vor allem in klinischen Studien verwendet und erlaubt eine histopathologische Beurteilung des Fibrosegrads, insbesondere im Übergangsstadium zwischen fortgeschrittener Fibrose und manifester Zirrhose:
- 0: keine Fibrose
- 1: Erweiterung der Portalfelder durch Fibrose
- 2: Portal-Fibrose mit wenigen kurzen Septen
- 3: Zahlreiche kurze Septen
- 4: Zahlreiche lange Septen mit partieller Läppchenzerstörung
- 5: inkomplette Zirrhose (fast vollständige Zerstörung)
- 6: manifeste Zirrhose
Ähnliche Scoringsysteme sind:
Ätiologie
Eine Leberzirrhose kann aufgrund verschiedener Grunderkrankungen entstehen. Die häufigsten Ursachen – bezogen auf die globalen Todesfälle durch Leberzirrhose – sind:
- Hepatitis C (HCV): 26,8 %
- Alkohol: 24,9 %
- Hepatitis B (HBV): 22,4 %
- nicht-alkoholische Fettlebererkrankung (NAFLD): 9,2 % (weltweit stark zunehmend)
- Sonstige: 16,7 %
Dabei gibt es deutliche regionale Unterschiede:
- Afrika und Ostasien: HBV bis zu 50–60 % aller Fälle, NAFLD noch relativ selten (< 10 %).
- Europa: Alkohol 40–60 %, HCV 10–15 %, HBV < 5 %, NAFLD zunehmend (5–15 %).
- Nordamerika: NAFLD stark zunehmend (bis 18 %), Alkohol 25–35 %, HCV 15–20 %.
- Südamerika: Gemischtes Bild, hohe HBV- und HCV-Prävalenz in bestimmten Regionen, Alkohol häufig.
- Tropische Regionen (z.B. Afrika, Südostasien): Bilharziose kann endemisch eine der Hauptursachen für portale Hypertension darstellen.
Alkoholische Lebererkrankung
Chronischer, übermäßiger Alkoholkonsum ist in Europa und Nordamerika die häufigste Ursache der Leberzirrhose. Ethanol und seine Metabolite (insbesondere Acetaldehyd) führen zu oxidativem Stress, Störung der mitochondrialen Funktion und einer proinflammatorischen Immunaktivierung. Es entwickelt sich eine alkoholische Fettleber, eine alkoholische Steatohepatitis und schließlich eine Fibrose und Zirrhose. Risikofaktor für eine alkoholische Lebererkrankung (ALD, ALE) ist ein täglicher Konsum von > 30 g Alkohol (Frauen) bzw. > 50 g (Männer) über mehrere Jahre, wobei genetische und metabolische Faktoren die Schwelle senken können.
Nicht-alkoholische Fettlebererkrankung
Die nicht-alkoholische Fettlebererkrankung ist mittlerweile eine der häufigsten chronischen Lebererkrankungen weltweit und spiegelt die Zunahme von Adipositas, Diabetes mellitus Typ 2 und metabolischem Syndrom wider. Insulinresistenz und viszerale Adipositas fördern die intrahepatische Lipidakkumulation und Lipotoxizität. Über eine einfache Steatose kommt es zu einer nicht-alkoholischen Steatohepatitis und schließlich zu Fibrose und Zirrhose.
Infektiöse Ursachen
Virushepatitiden sind eine häufige Ursache der Leberzirrhose:
- Chronische HBV-Infektionen führen über eine anhaltende Immunreaktion und direkte zytopathische Effekte zu einer progredienten Fibrose. Besonders hohe Risiken bestehen bei Koinfektionen mit HDV oder HIV.
- Persistierende HCV-Infektionen verursachen eine kontinuierliche entzündliche Aktivität, die über Jahrzehnte zur Zirrhose fortschreiten kann. Seit der Einführung direkter antiviraler Therapien (DAA) ist die HCV-assoziierte Zirrhose in vielen Regionen rückläufig.
- Hepatitis D ist nur in Kombination mit HBV möglich und führt zu einer besonders aggressiven Verlaufsform.
- Hepatitis E: selten chronisch verlaufend, insbesondere bei immunsupprimierten Patienten.
Neben viralen Hepatitiden können auch andere Infektionen eine Zirrhose verursachen:
- Bilharziose (Schistosomiasis): In tropischen Regionen eine häufige Ursache portaler Hypertension. Parasitäre Ablagerungen in der Pfortaderregion führen zu einer periportalen Fibrose, die in eine Zirrhose mündet.
- Tuberkulose, Syphilis: selten, meist sekundär.
- Chronische bakterielle Cholangitiden: z.B. bei Gallenwegsstenosen.
Autoimmunerkrankungen der Leber
Hierzu zählen vor allem:
- Autoimmunhepatitis (AIH): Immunvermittelte chronische Entzündung, die unbehandelt zu fortschreitender Fibrose und Zirrhose führt.
- Primär biliäre Cholangitis (PBC): Chronische Zerstörung der intrahepatischen kleinen Gallengänge, charakteristisch sind antimitochondriale Antikörper (AMA).
- Primär sklerosierende Cholangitis (PSC): Entzündlich-fibrosierende Erkrankung der intra- und extrahepatischen Gallenwege, häufig assoziiert mit chronisch-entzündlichen Darmerkrankungen (z. B. Colitis ulcerosa).
Metabolische und genetische Erkrankungen
- Hämochromatose: Eisenüberladung führt zu oxidativem Stress und direkten Gewebeschäden.
- Morbus Wilson: Kupferspeicherung mit toxischen Effekten auf Hepatozyten.
- Alpha-1-Antitrypsin-Mangel: Fehlgefaltetes Protein lagert sich in Hepatozyten ab und führt zu Zellschädigung.
- Nicht-zirrhotische Speichererkrankungen wie Glykogenspeicherkrankheiten können bei Progression ebenfalls in eine Zirrhose übergehen.
- Mukoviszidose (zystische Fibrose): Gallengangsobstruktion durch zähes Sekret mit sekundärer biliärer Zirrhose.
Biliäre und vaskuläre Ursachen
- Chronische Gallenwegsobstruktionen, z.B. durch Gallensteine, Strikturen oder Tumoren, können sekundär eine biliäre Zirrhose verursachen
- Budd-Chiari-Syndrom (Thrombose der Lebervenen)
- Chronisches sinusoidales Obstruktionssyndrom (venöse okklusive Leberkrankheit, VOD): z.B. nach Chemotherapie oder Knochenmarktransplantation)
- Pfortaderthrombose (indirekt durch Minderperfusion und sekundäre Fibrose)
Kardiale Ursachen
Eine chronische Rechtsherzinsuffizienz oder konstante Stauung der Lebervenen kann zu einer sogenannten kardialen Zirrhose (Cirrhose cardiaque) führen. Sie tritt häufig bei Patienten mit schwerer Trikuspidalinsuffizienz oder konstriktiver Perikarditis auf.
Medikamentös-toxische Ursachen
Langfristige Exposition gegenüber hepatotoxischen Substanzen wie Aflatoxin B1, Vinylchlorid, Thallium oder Arsen kann zur chronischen Leberschädigung führen. Auch bestimmte Medikamente können bei chronischer Einnahme eine progrediente Hepatitis mit nachfolgender Fibrose verursachen (z.B. Amiodaron, Methyldopa, Isoniazid, Valproat oder langfristig höherdosiertes Methotrexat).
Kryptogene Zirrhose
In etwa 5–10 % der Fälle kann trotz umfassender Diagnostik keine eindeutige Ursache identifiziert werden. Häufig handelt es sich retrospektiv um eine fortgeschrittene NAFLD, bei der typische Merkmale durch den Umbauprozess nicht mehr erkennbar sind.
Pathogenese und Pathophysiologie
Die Leberzirrhose entsteht als Endstadium fortgesetzter chronischer Leberschädigung, unabhängig von der initialen Ursache. Gemeinsam ist allen Formen ein Prozess aus persistierender Entzündung, Aktivierung von Fibrosemechanismen, vaskulärem Remodeling und Verlust der physiologischen Gewebearchitektur.
Initiale Schädigung und Zelluntergang
Am Beginn steht eine primäre Schädigung von Hepatozyten oder intrahepatischen Gallengangsepithelien durch Noxen (z.B. Alkohol). Es kommt zu einer Nekrose und Apoptose von Hepatozyten mit Freisetzung von intrazellulären DAMPs (damage-associated molecular patterns). Nach Aktivierung von Kupffer-Zellen, dendritischen Zellen und neutrophilen Granulozyten kommt es zur Freisetzung von proinflammatorischen Zytokinen (TNF, IL-1β, IL-6). Die chronische Entzündung bleibt bestehen, wenn die Noxe fortbesteht oder die Regeneration nicht ausreicht.
Aktivierung der hepatischen Sternzellen
Die zentralen Effektorzellen der Fibrogenese sind die hepatischen Sternzellen (HSC, Ito-Zellen), die sich im Disse-Raum zwischen Hepatozyten und Sinusoiden befinden. Im Ruhezustand speichern HSC Vitamin A und sind metabolisch inaktiv. Durch entzündliche Signale, oxidativen Stress, TGF-β und PDGF werden sie aktiviert und in myofibroblastenartige Zellen umgewandelt. Aktivierte HSC produzieren große Mengen extrazellulärer Matrix (ECM), insbesondere Kollagen Typ I und III, und erhöhen den intrahepatischen Widerstand durch Vasokonstriktoren (z.B. Endothelin-1).
Durotaxis
Ein Schlüsselmechanismus der Fibroseprogression ist die Durotaxis, die gerichtete Migration von Zellen entlang eines Steifigkeitsgradienten des Gewebes. Lokale Fibroseherde erhöhen die Gewebesteifigkeit in umschriebenen Arealen. HSC besitzen Integrine und Signalwege wie YAP/TAZ, die mechanische Spannung erkennen. Je steifer die Umgebung, desto stärker werden HSC aktiviert und wandern gezielt in diese Areale ein. Dort produzieren sie weiter Kollagen und ECM, sodass ein Circulus vitiosus entsteht, in dem die Fibrose ab einem kritischen Punkt exponentiell fortschreitet.
Progression der Fibrose
Die fortschreitende Fibrosierung führt zu einem irreversiblen Umbau der normalen Leberarchitektur:
- Bildung von Brückenfibrosen: Kollagene Septen verbinden die Portalfelder miteinander oder mit Zentralvenen.
- Regeneratknoten: Verbleibende vitale Hepatozyten proliferieren in umschlossenen Arealen und bilden knotige Strukturen.
- Störung des Sinusoiden: Verlust der Fenestrierung der Sinusendothelien ("Kapillarisation"), wodurch der Austausch zwischen Blut und Hepatozyten eingeschränkt wird.
- Vaskuläres Remodeling: Blutflüsse werden umgelenkt, porto-systemische Shunts entstehen.
Entwicklung der portalen Hypertension
Die portale Hypertension ist eine direkte Folge der architektonischen Zerstörung und der dynamischen Vasokonstriktion innerhalb der Leber:
- Mechanische Komponente: Kollagene Septen und Regeneratknoten verengen die Sinusoide und blockieren den Blutfluss.
- Dynamische Komponente: Aktivierte HSC und Endothelzellen produzieren Vasokonstriktoren (v.a. Endothelin-1) und vermindern vasodilatierende Faktoren (z.B. NO)
Die Folge sind Umgehungskreisläufe (Shunts) zwischen Pfortader und systemischem Kreislauf. Diese portokavale Anastomosen leiten das Blut an der Leber vorbei in gastroösophageale und rektale Venen sowie in Venen der Bauchwand um.
Systemische Kreislaufveränderungen
Die portale Hypertension führt zu einer Hyperämie im Splanchnikusgebiet und systemischen Kreislaufstörungen:
- Vasodilatation der Mesenterialgefäße durch vermehrtes NO und andere Mediatoren: Abfall des systemischen Gefäßwiderstands.
- Aktivierung von RAAS, Sympathikus und Vasopressin: Natrium- und Wasserretention und damit Aszites, sekundärer Hyperaldosteronismus und hepatorenales Syndrom (HRS).
Langfristig entsteht eine hyperdyname Kreislaufsituation mit hohem Herzzeitvolumen und niedrigen systemischen Widerständen.
Leberinsuffizienz
Parallel zur portalen Hypertension entwickelt sich eine funktionelle Einschränkung der Leber durch Verlust funktionsfähigen Parenchyms:
- Verminderte Syntheseleistung: Hypoalbuminämie, Gerinnungsstörung
- Reduzierte Entgiftungskapazität: Anstieg von Ammoniak und anderen Neurotoxinen führen zur hepatischen Enzephalopathie
- Störung der Immunfunktion: erhöhte Infektanfälligkeit (z.B. spontan bakterielle Peritonitis)
Dynamik der Fibrose
Bei erfolgreicher Behandlung der Grunderkrankung können fibrotische Strukturen teilweise regredieren. Aktivierte HSC können in einen Ruhezustand zurückkehren oder apoptotisch eliminiert werden. Bei fortbestehender Noxe schreitet der Prozess fort, was zu einem irreversiblen Umbau mit dominierenden Regeneratknoten und breiten Septen führt.
Akut-auf-chronisches Leberversagen
Bei fortgeschrittener Zirrhose kann ein akuter Auslöser (Infektion, Alkohol, Blutung, Medikamente) ein ACLF verursachen. Die massive Freisetzung proinflammatorischer Zytokine ("Zytokinsturm") bedingt dann ein Multiorganversagen. Pathophysiologisch spielt eine systemische Inflammation und Mikrozirkulationsstörung eine zentrale Rolle.
Organsyndrome
Die Leberzirrhose wirkt sich nicht nur auf das Leberparenchym selbst aus, sondern führt auch zu systemischen Funktionsstörungen anderer Organe. Diese resultieren aus der Kombination von portaler Hypertension, systemischer Vasodilatation, neurohumoraler Aktivierung, systemischer Inflammation und der Akkumulation toxischer Metabolite, die infolge portosystemischer Shunts nicht mehr von der Leber eliminiert werden. Die wichtigsten Syndrome sind das hepatorenale Syndrom, das hepatopulmonale Syndrom, die portopulmonale Hypertonie, die hepatische Enzephalopathie sowie das hepatokardiale Syndrom.
Hepatorenales Syndrom
Das hepatorenale Syndrom ist eine funktionelle Nierenfunktionsstörung, die bei fortgeschrittener Zirrhose und ausgeprägter portaler Hypertension auftritt. Es liegt keine primäre strukturelle Nierenschädigung vor, sondern eine renale Vasokonstriktion. Die Pathogenese beginnt mit einer starken Vasodilatation im Splanchnikusgebiet (s.o.). Diese führt zu einem relativen Volumenmangel im Kreislauf und aktiviert RAAS, Sympathikus und Vasopressin. Die Folge ist eine kompensatorische Vasokonstriktion der Nierengefäße, welche die GFR massiv senkt.
Hepatopulmonales Syndrom
Das hepatopulmonale Syndrom (HPS) ist charakterisiert durch die Trias aus Leberzirrhose, portaler Hypertension und arterieller Hypoxämie. Ursache ist eine intrapulmonale Gefäßerweiterung, die eine pathologische Durchmischung von oxygeniertem und nicht-oxygeniertem Blut verursachen (Shuntbildung). NO und andere vasodilatierende Mediatoren aus dem Kreislauf gelangen über portosystemische Shunts in die Lunge und führen dort zu einer inadäquaten Gefäßerweiterung.
Portopulmonale Hypertonie
Die portopulmonale Hypertonie (PoPH) ist eine Form der präkapillären pulmonalen Hypertonie. Anders als beim HPS steht hier nicht eine Vasodilatation, sondern die Vasokonstriktion und das Remodeling der Pulmonalgefäße im Vordergrund. Die Folge ist ein erhöhter pulmonaler Gefäßwiderstand, der zur Rechtsherzbelastung und im Endstadium zu einer Rechtsherzinsuffizienz führt.
Hepatische Enzephalopathie
Die hepatische Enzephalopathie beschreibt ein kontinuierliches Spektrum neuropsychiatrischer Funktionsstörungen, das von diskreten kognitiven Defiziten bis zum Coma hepaticum reicht. Pathophysiologisch spielen Ammoniak und andere neurotoxische Substanzen eine Schlüsselrolle, die aufgrund portosystemischer Shunts und eingeschränkter Leberfunktion nicht mehr eliminiert werden. Im Gehirn führt Ammoniak zur Astrozytenschwellung (Alzheimer-Typ-II-Astrozyten) und zu Veränderungen der Neurotransmission, verstärkt durch systemische Entzündung und erhöhte Mangan-Deposition. Auslöser sind häufig Infektionen, gastrointestinale Blutungen oder Elektrolytstörungen.
Hepatokardiales Syndrom
Das hepatokardiale Syndrom beschreibt eine strukturelle und funktionelle Herzmuskelerkrankung bei fortgeschrittener Leberzirrhose ohne primäre Herzerkrankung. Chronische Hyperzirkulation und anhaltende Exposition gegenüber proinflammatorischen Mediatoren führen zu Myokardhypertrophie, Fibrose und Elektrolytstörungen.
Weitere Syndrome
Neben den klassischen hepatischen Organsyndromen gibt es weitere Funktionsstörungen, die im Rahmen einer Zirrhose auftreten können:
- Hepato-adrenales Syndrom: Nebenniereninsuffizienz bei fortgeschrittener Lebererkrankung mit Hypotonie und erhöhter Mortalität.
- Hepato-thyreoidales Syndrom: Low-T3-Syndrom als Ausdruck des katabolen Stoffwechsels, prognostisch ungünstig.
- Hepato-hämatologisches Syndrom: Hypersplenismus mit Anämie, Leukopenie und Thrombozytopenie; verstärkt Gerinnungsstörungen.
- Hepato-muskuläres Syndrom: Sarkopenie und Muskelabbau als Folge von Hypermetabolismus und Mangelernährung, starker Prädiktor für Transplantationsrisiko.
Pathologie
Makroskopie
Makroskopisch zeigt die zirrhotische Leber eine unregelmäßige, höckerige Oberfläche, die durch Regeneratknoten und breite Fibrosesepten geprägt ist. Die Konsistenz ist derb und fest, das Gewicht in fortgeschrittenen Stadien meist reduziert. In frühen Stadien kann die Leber jedoch normal groß oder sogar vergrößert sein, insbesondere bei gleichzeitiger Fettleber oder entzündlicher Aktivität.
Die Farbe der Leber variiert je nach Grunderkrankung:
- Alkohol- oder NAFLD-bedingte Zirrhosen sind häufig gelblich verfärbt durch Steatose.
- Bei cholestatischen Formen (z. B. PBC, PSC) zeigt sich eine grünliche Verfärbung durch Gallenstau.
- Dunkelbraune Tönung tritt bei Hämochromatose auf (Eisenablagerung), kupferrote bis orangefarbene Farbtöne sind bei Morbus Wilson typisch.
Nach der Größe der Regeneratknoten unterscheidet man zwischen mikronodulärer, makronodulärer und gemischt nodulärer Zirrhose (s.o.)
Die Verteilung der Fibrose kann Hinweise auf die Ätiologie geben:
- Bei Alkohol- oder Virus-bedingten Zirrhosen sind Brückenfibrosen zwischen Portalfeldern und Zentralvenen typisch.
- Cholestatische Zirrhosen beginnen bevorzugt periportal.
- Bei kardialer Stauungszirrhose liegt der Schwerpunkt zentrolobulär ("Muskatnussleber").
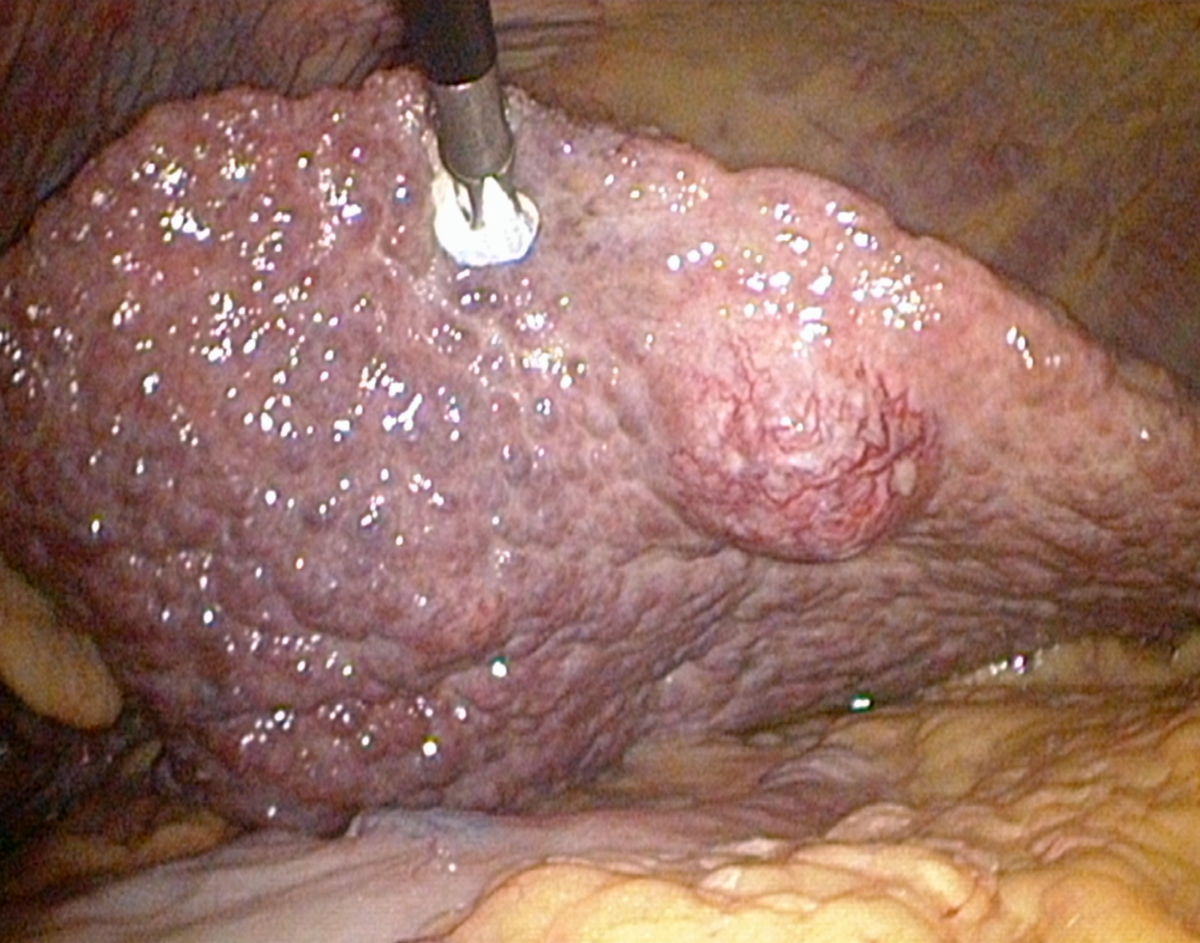
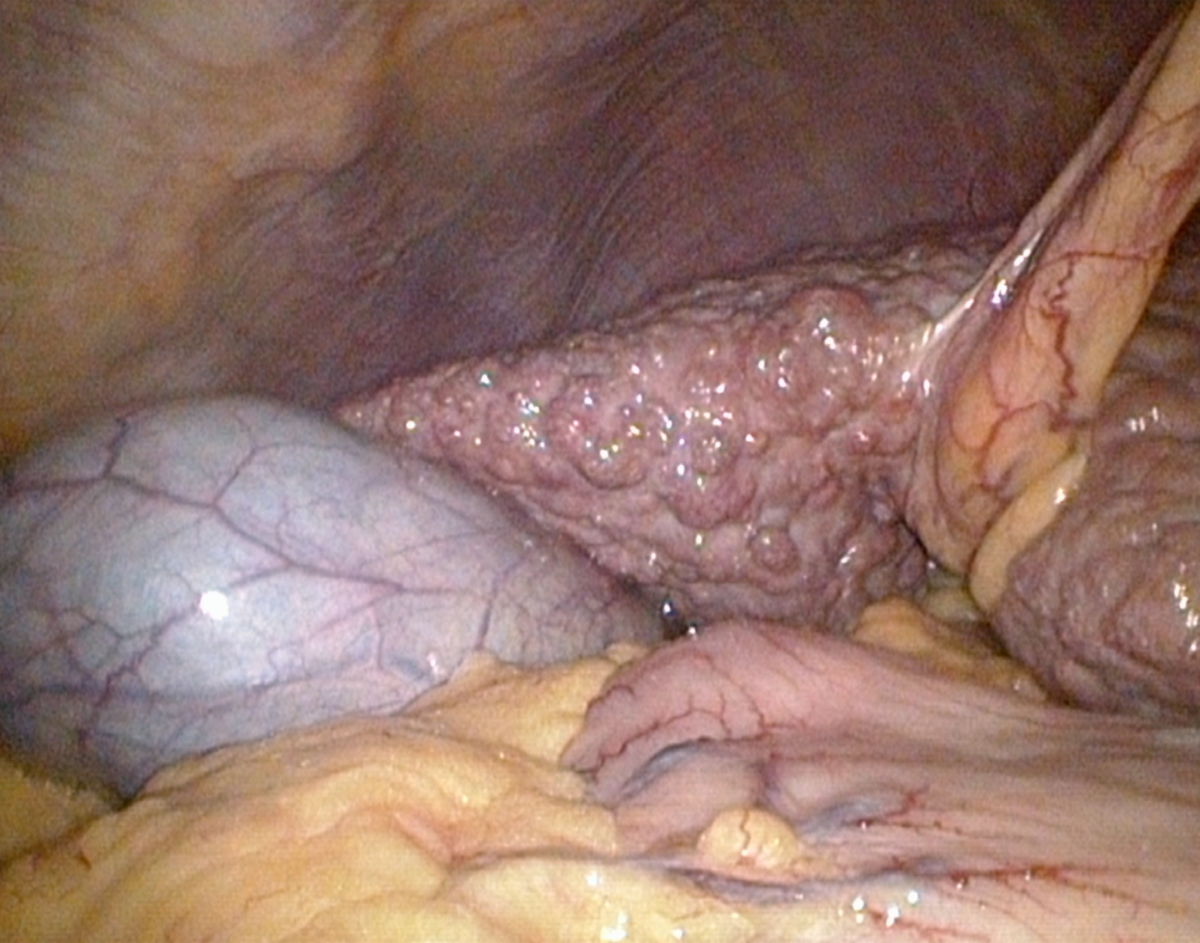
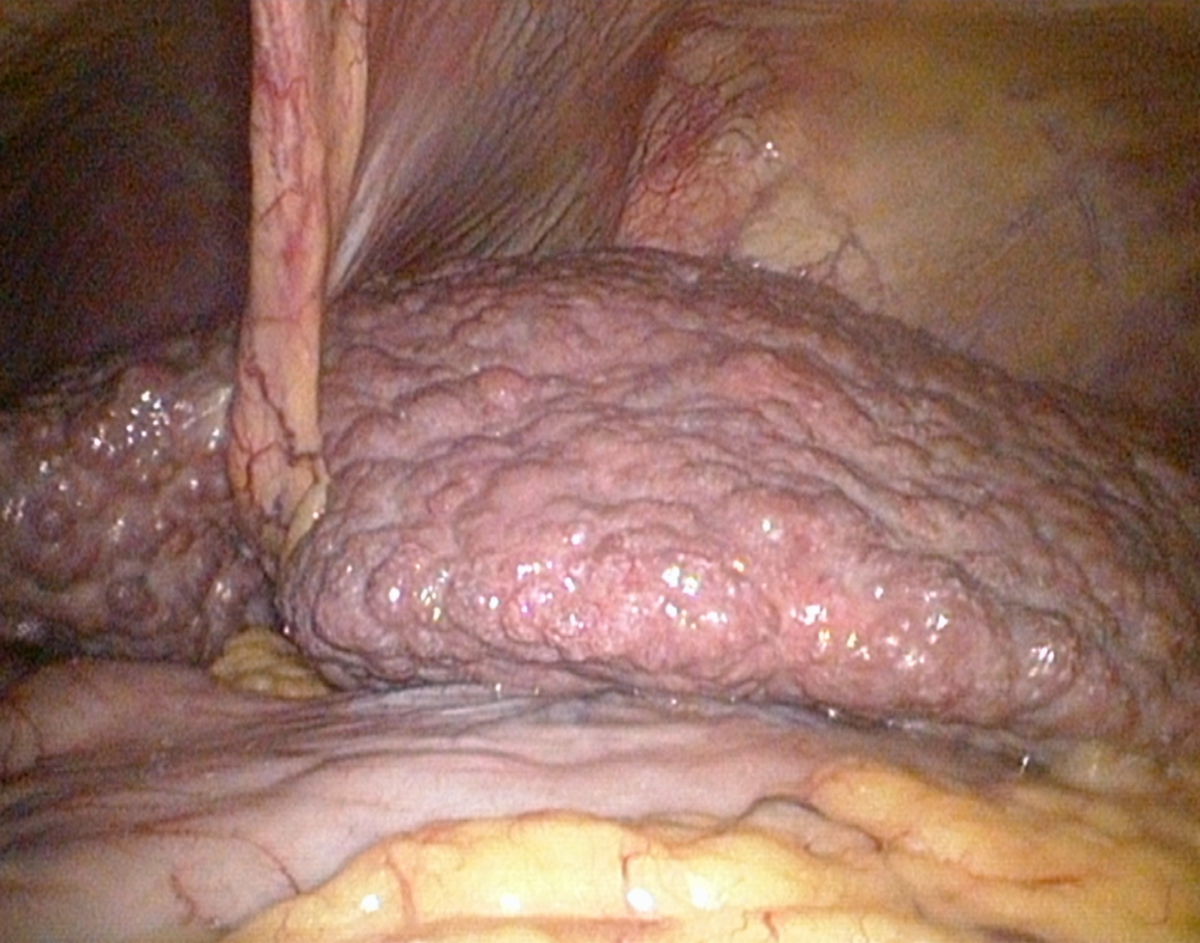
Pathohistologie
Histologisch ist die Zirrhose durch das Nebeneinander von Regeneration und Destruktion gekennzeichnet. Merkmale sind:
- Regeneratknoten: Diese bestehen aus vitalen, proliferierenden Hepatozyten. Sie sind vollständig von breiten fibrotischen Septen umgeben, sodass kein geordneter Blut- oder Gallefluss mehr möglich ist. Die Regeneratknoten sind unregelmäßig geformt, unterschiedlich groß und enthalten häufig gestörte Läppchenstrukturen.
- Fibrosesepten: Die Septen bestehen überwiegend aus Kollagen I und III, produziert von aktivierten hepatischen Sternzellen und myofibroblastischen Zellen. Sie verbinden Portalfelder mit Zentralvenen (Brückenfibrose) und bilden ein unregelmäßiges Bindegewebsnetzwerk, das die ursprüngliche Läppchenstruktur vollständig zerstört.
- Sinusoidale Veränderungen: Die Sinusoide verlieren ihre typische Fenestrierung ("Kapillarisation"), wodurch der Stoffaustausch zwischen Blut und Hepatozyten massiv eingeschränkt wird. Gleichzeitig entstehen durch Neoangiogenese neue Gefäßverbindungen innerhalb der Septen, die direkt in systemische Venen münden. Dies führt zur Ausbildung intrahepatischer porto-systemischer Shunts, die den Funktionsverlust weiter verstärken.
- Hepatozytenveränderungen:
- Alkoholische Zirrhose: ballonierte Zellen mit Mallory-Denk-Körperchen und makrovesikulärer Steatose.
- NAFLD/NASH: makro- und mikrovesikuläre Steatose, entzündliche Infiltrate
- Morbus Wilson: Kupferablagerungen, sichtbar in Spezialfärbungen (Rhodanin, Orcein)
- Hämochromatose: intrazelluläre Eisenablagerungen, nachweisbar in Perls-Färbung
- Autoimmunhepatitis: dichte lymphoplasmazelluläre Infiltrate, oft periportal
- Gallengangsveränderungen: Bei cholestatischen Formen wie der PBC oder PSC kommt es zur Destruktion intrahepatischer Gallengänge und zu typischer zwiebelschalenartiger Fibrose um die betroffenen Duktusstrukturen. Gallenstau führt zu gelbbraunen Pigmentablagerungen in Hepatozyten und Kupffer-Zellen.
- Entzündungsaktivität: Die Intensität der Entzündung hängt von der Grunderkrankung und dem Stadium ab. Bei viralen und autoimmunen Ursachen findet sich eine ausgeprägte lymphozytäre Infiltration, während in späten, inaktiven Stadien die Entzündung zurücktritt und die Fibrose dominiert. In alkoholischen Zirrhosen oder bakteriellen Superinfektionen sind neutrophile Granulozyten prominent.
Spezielle Muster:
- Bei kardialer Zirrhose stehen zentrolobuläre Nekrosen mit begleitender Fibrose um die Zentralvenen im Vordergrund.
- Toxische Ursachen führen häufig zu makronodulären Umbauten und sind mit einem erhöhten Risiko für Angiosarkome assoziiert.
- Metall-Speichererkrankungen wie Hämochromatose oder Morbus Wilson zeigen typische Ablagerungsmuster, die sich immunhistochemisch darstellen lassen.
Symptome
Die Symptomatik der Leberzirrhose hängt stark vom Krankheitsstadium ab. In frühen Stadien bleibt die Erkrankung häufig lange asymptomatisch oder zeigt nur unspezifische Allgemeinzeichen. Erst mit dem Übergang in die Dekompensation treten die typischen Manifestationen auf, die durch portale Hypertension, Leberinsuffizienz und systemische Komplikationen verursacht werden.
Kompensierte Zirrhose
In der kompensierten Phase sind die klinischen Zeichen oft diskret, sodass die Diagnose häufig zufällig gestellt wird, etwa durch Laboruntersuchungen oder Bildgebung:
- Unspezifische Allgemeinsymptome: Müdigkeit, Abgeschlagenheit, Leistungsabfall, Druck im rechten Oberbauch, Meteorismus, Übelkeit, Gewichtsabnahme
- Frühe Leberzeichen: gelegentlich leichte Hepatomegalie oder Splenomegalie ohne Aszites oder Ikterus.
In diesem Stadium liegt die 1-Jahres-Überlebensrate meist über 90 %, insbesondere bei Child-Pugh A.
Dekompensierte Zirrhose
Die Dekompensation markiert einen entscheidenden Wendepunkt mit deutlichem Abfall der Überlebenswahrscheinlichkeit. Typische Symptome und Zeichen:
- Aszites: Häufigstes und oft erstes Zeichen der Dekompensation. Klinisch erkennbar durch Bauchumfangszunahme, Flankenauswölbung und Dämpfung bei Perkussion.
- Ikterus: Folge der verminderten hepatischen Bilirubinkonjugation und -ausscheidung. Zunächst Sklerenikterus, später generalisierte Gelbfärbung der Haut. Häufig begleitet von Juckreiz (v.a. bei cholestatischen Ursachen).
- Hepatische Enzephalopathie: Frühsymptome sind Schlaf-Wach-Rhythmusstörungen, Reizbarkeit, kognitive Verlangsamung. Später Asterixis ("Flapping Tremor"), Desorientierung, Somnolenz bis hin zum Coma hepaticum.
- Varizenblutung: Klinisch manifest durch Hämatemesis, Teerstuhl oder Kreislaufkollaps. Notfall mit hoher Letalität, erfordert sofortige interventionelle Therapie.
- Hepatorenales Syndrom: Oligurie, steigendes Kreatinin ohne strukturelle Nierenschädigung.
Spezifische Haut- und Allgemeinzeichen
Mit zunehmender Leberinsuffizienz treten charakteristische Haut- und Systemmanifestationen auf, die als "Leberzeichen" bekannt sind:
- Spider naevi: sternförmige, rötliche Gefäßneubildungen mit zentralem arteriellen Punkt, v.a. im oberen Thoraxbereich, Gesicht und Hals
- Teleangiektasien: feine, sichtbare Erweiterungen kleiner Blutgefäße, nicht nur in typischen Spider naevi, sondern auch diffus verteilt
- Caput medusae: sichtbare, geschlängelte Bauchwandvenen bei portaler Hypertension
- Palmarerythem: symmetrische Rötung der Thenar- und Hypothenarregion
- Plantarerythem: flächige Rötung der Fußsohlen, analog zum Palmarerythem
- Lacklippen: stark gerötete, glänzende Lippen durch gesteigerte Durchblutung
- Lackzunge: intensiv gerötete, glatte und glänzende Zunge, typischer Ausdruck der Hyperzirkulation und Hypovitaminose
- Mundwinkelrhagaden: schmerzhafte, teils infizierte Einrisse der Mundwinkel infolge Vitamin-B-Mangel und Mangelernährung
- Weißnägel (Leukonychie) oder Terry-Nägel
- Prurigo simplex subacuta: juckende, papulöse Hautläsionen durch chronischen Pruritus bei Cholestase
- Hautatrophie: dünne, pergamentartige Haut infolge Hypoalbuminämie und gestörter Kollagensynthese
- Dupuytren-Kontraktur: Bindegewebsstrangbildungen in der Palmaraponeurose, besonders häufig bei alkoholischer Zirrhose
- Hyperpigmentierung: bräunliche Hautverfärbung bei Hämochromatose ("Bronzediabetes") oder generalisiert bei chronischer Leberinsuffizienz
- Foetor hepaticus: süßlich-muffiger Atemgeruch durch Dimethylsulfid und andere flüchtige Substanzen
Extrahepatische Manifestationen
Leberzirrhose ist eine systemische Erkrankung. Häufig finden sich Symptome durch Beteiligung anderer Organe:
- Kardiovaskulär: Zeichen der Hyperzirkulation, Tachykardie, niedriger Blutdruck, Belastungsdyspnoe bei hepatokardialem Syndrom
- Pulmonal: Dyspnoe bei hepatopulmonalem Syndrom oder portopulmonaler Hypertonie
- Neurologisch: kognitive Störungen bis zum Koma bei Enzephalopathie
- Muskuloskelettal: Sarkopenie durch Hyperkatabolismus
- Endokrinologisch: Gynäkomastie, Hodenatrophie, Libidoverlust, Amenorrhö, Potenzstörungen, Hypothyreose, Bauchglatze
Komplikationen
Die häufigsten Komplikationen der Leberzirrhose entstehen durch portale Hypertension und Leberinsuffizienz und markieren den Übergang zur dekompensierten Erkrankung. Die wichtigsten Komplikationen sind:
- Aszites mit spontan bakterieller Peritonitis (SBP): Fieber, diffuse Bauchschmerzen
- Varizenblutungen: z.B. mit Hämatemesis und hypovolämischem Schock. Eine Ösophagusvarizenblutung hat eine Letalität von bis zu 30 %.
- hepatische Enzephalopathie: mit neuropsychiatrischen Symptome bis hin zum Coma hepaticum
- Hepatorenales Syndrom mit funktionellen Nierenversagen
Eine besonders schwere Verlaufsform ist das Akut-auf-chronische Leberversagen (ACLF). Dabei kommt es nach einem auslösenden Ereignis wie Infektion oder Blutung zu einer raschen systemischen Entzündungsreaktion und Multiorganversagen. ACLF ist mit einer sehr hohen Kurzzeitmortalität verbunden und erfordert sofortige intensivmedizinische Betreuung und die rasche Prüfung einer Transplantationsindikation.
Langfristig erhöht die Zirrhose das Risiko für ein hepatozelluläres Karzinom (HCC), das oft asymptomatisch verläuft und daher nur durch regelmäßiges Screening frühzeitig erkannt werden kann.
Diagnostik
Die Diagnostik der Leberzirrhose umfasst die Erkennung der Erkrankung, die Abklärung der Ätiologie, die Einschätzung des Schweregrades sowie das Monitoring auf Komplikationen. Sie stützt sich auf körperliche Untersuchung, Laboruntersuchungen, bildgebende Verfahren und ggf. invasive Methoden.
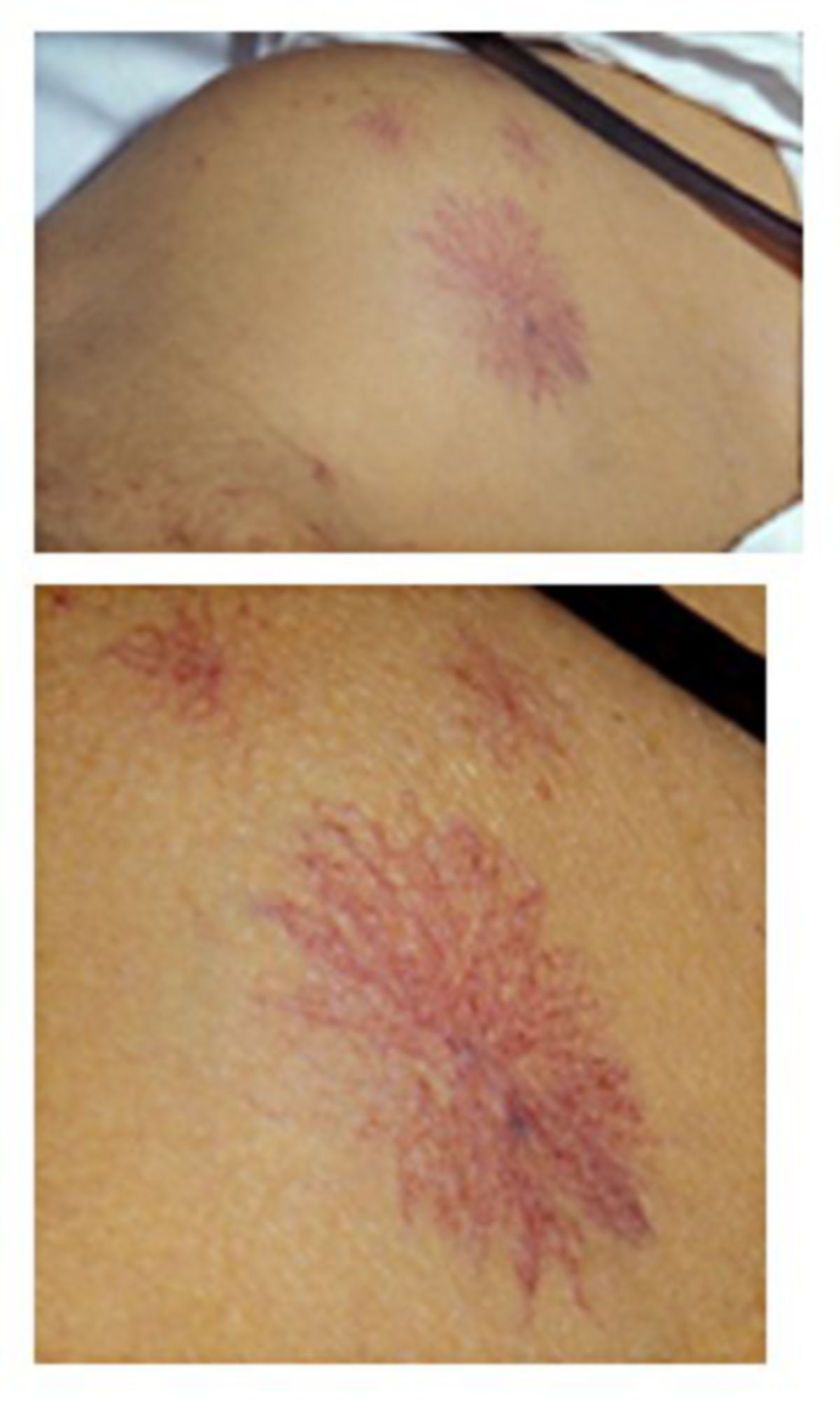
Körperliche Untersuchung
Die Diagnose kann oft schon anhand der Anamnese und körperlichen Untersuchung vermutet werden.
- Anamnese: Alkohol- oder Medikamentenkonsum, Virushepatitis, Stoffwechsel- oder Autoimmunerkrankungen, Symptome wie Müdigkeit, Pruritus, Aszites oder gastrointestinale Blutungen
- Hepatomegalie oder kleine, harte "Schrumpfleber"
- Splenomegalie als Hinweis auf portale Hypertension
- Typische Leberzeichen (z.B. Spider naevi, Gynäkomastie, Aszites)
- Zeichen einer Dekompensation: Ikterus, Ödeme, Bewusstseinsstörungen
Labor
Laborwerte geben Aufschluss über Schweregrad, Funktionsverlust und die mögliche Ätiologie:
- Leberfunktionsparameter:
- Bilirubin: erhöht bei cholestatischer Komponente oder starker Hepatozytenschädigung.
- Albumin: erniedrigt bei fortgeschrittener Synthesestörung.
- INR/Quick: verlängerte Gerinnungszeit durch Mangel an Gerinnungsfaktoren.
- Cholinesterase: erniedrigt als Marker der Syntheseleistung.
- Zellschädigungsmarker:
- Knochenmark/Splenomegalie:
- Thrombozytopenie, Leukopenie, Anämie durch Hypersplenismus.
- Ätiologische Abklärung:
- Hepatitis-Serologie (HBV, HCV).
- Autoantikörper (ANA, AMA, SMA) bei Autoimmunerkrankungen.
- Ferritin, Transferrinsättigung (Hämochromatose).
- Coeruloplasmin, Kupfer im Serum und 24h-Urin (Morbus Wilson).
- Alpha-1-Antitrypsin-Spiegel
Laborparameter sind auch Grundlage für Scores wie Child-Pugh und MELD zur Prognoseeinschätzung.
| Erniedrigt | Erhöht |
|---|---|
| Cholinesterase | γ-Globulinfraktion (Proteinelektrophorese) |
| Vitamin K-abhängige Gerinnungsfaktoren | Ammoniak (im Spätstadium) |
| Antithrombin III, Albumin im Serum | AST, ALT, GLDH und γ-GT (im entzündlichen Schub) |
| Thrombozyten bei Splenomegalie | AP, LAP, γ-GT und evtl. Bilirubin (bei Cholestase) |
| Natrium (Hyponatriämie), Kalium (Hypokaliämie) |
Bildgebung
Bildgebende Verfahren spielen eine zentrale Rolle in der Diagnosesicherung, im Staging und im Monitoring.
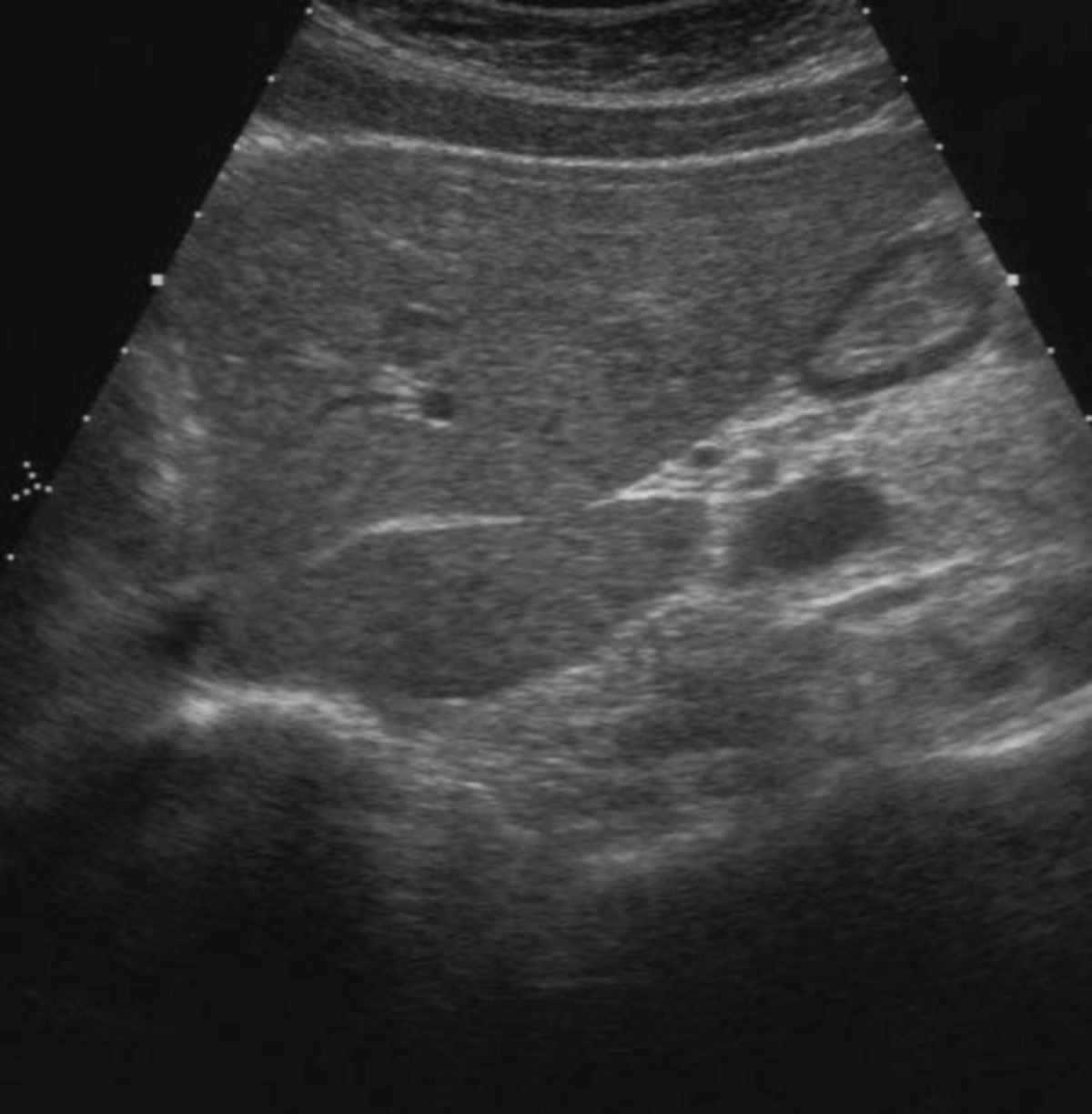
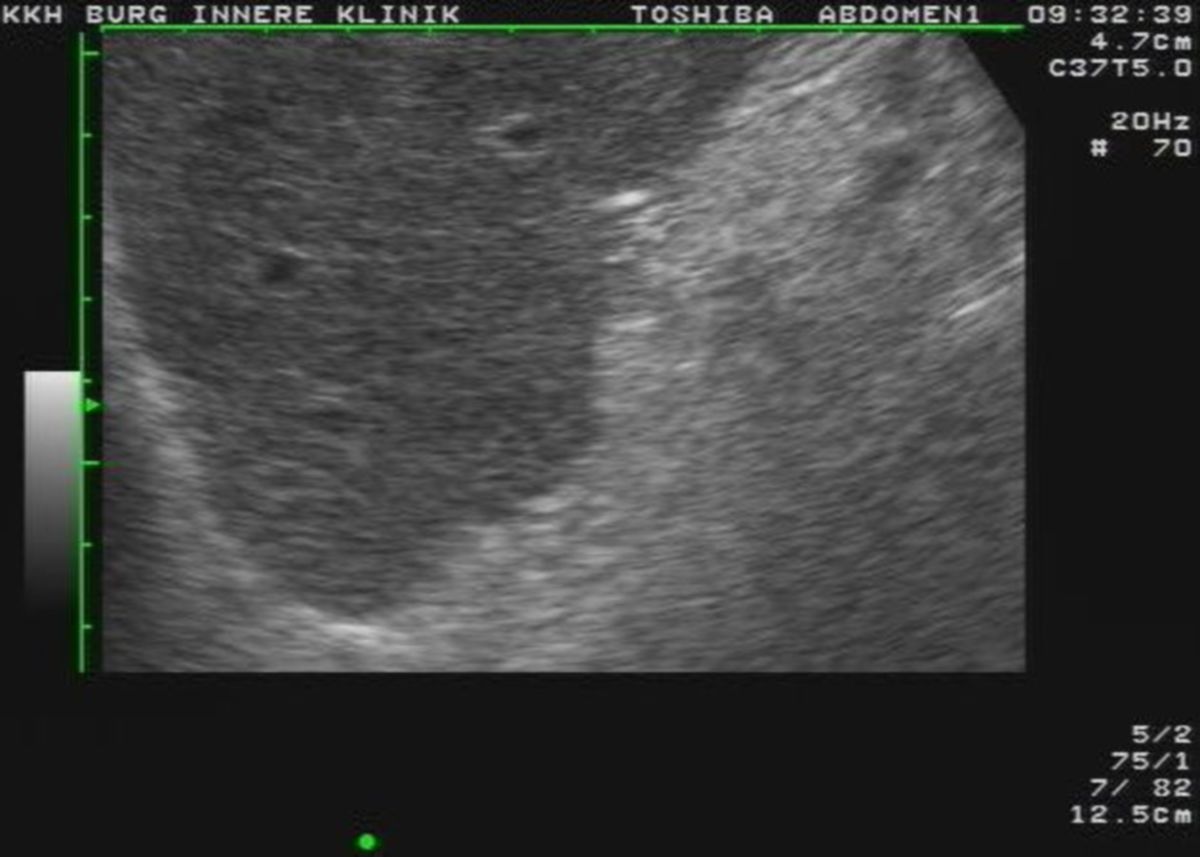
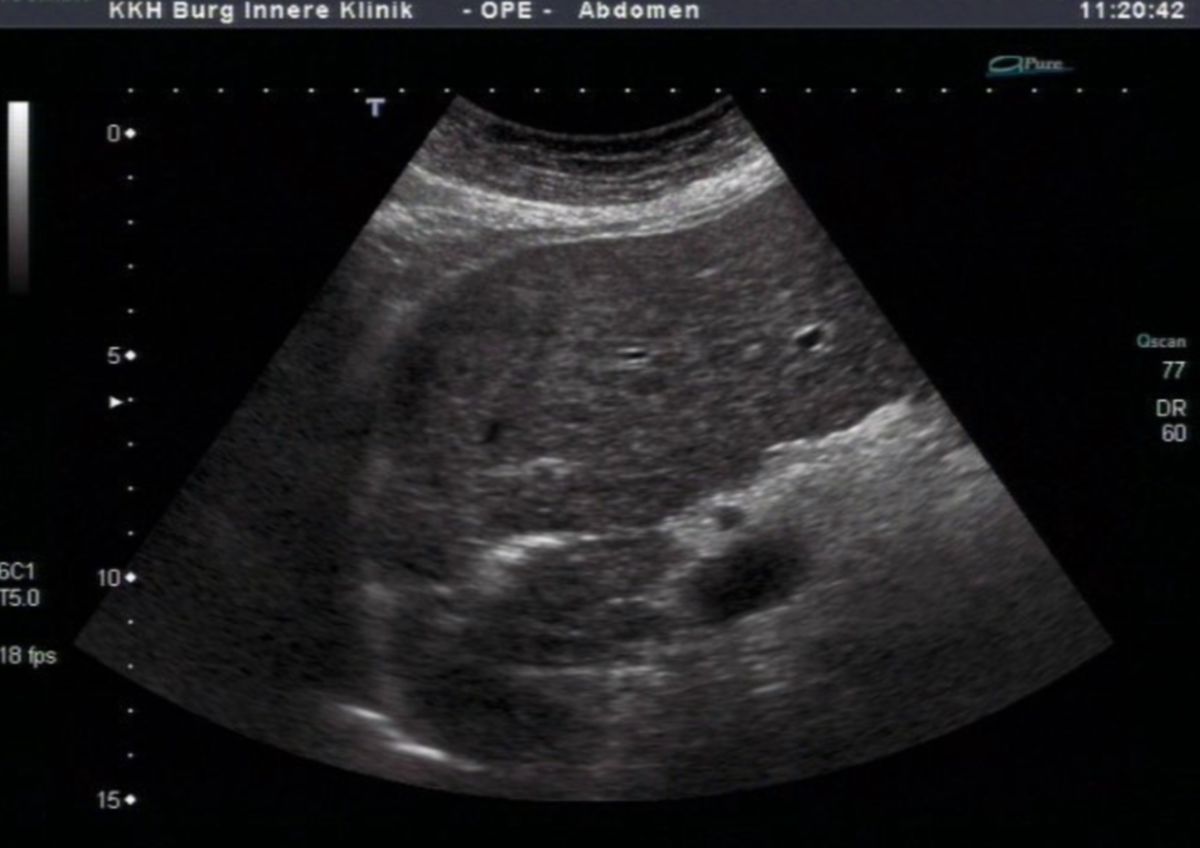
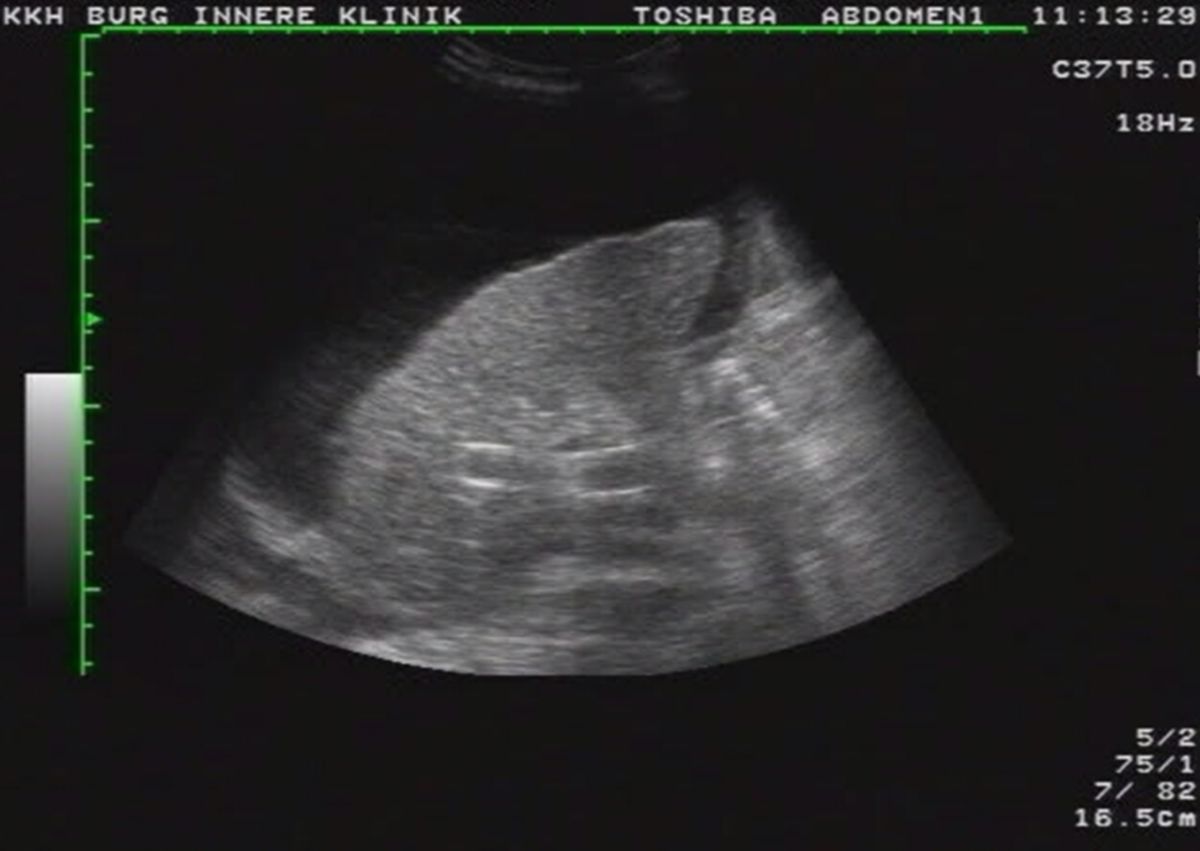
Sonographie
Die Sonographie ist die bildgebende Methode der ersten Wahl. Die wichtigsten Veränderungen, die sich in der Sonographie nachweisen lassen, sind:
- unregelmäßige, höckrige Leberoberfläche mit abgerundeten Rändern
- inhomogenes Leberparenchym
- Regeneratknötchen
- verminderte Lebervenen
- gestaute Kollateralkreisläufe bzw. Varizen
- Aszites
- Splenomegalie
- Screening auf HCC (alle 6 Monate)
Transiente Elastographie
Ein von der Sonographie abgeleitetes Verfahren zur Messung des Bindegewebsanteils der Leber ist die transiente Elastographie (Fibroscan®). Die Lebersteifigkeit korreliert mit Fibrosegrad. Ein Wert von 12–14 kPa spricht stark für eine Zirrhose.
Computertomographie
In der Computertomographie ist eine detaillierte morphologische Beurteilung möglich. Sie ist indiziert
- wenn Ultraschallbefunde unklar sind oder für HCC-Screening eine weitergehende Abklärung notwendig ist,
- bei akuten Komplikationen, z.B. Varizenblutung, Perforation, Thrombose der Venae portae oder Lebervenen,
- zur präoperativen Planung vor Leberresektion oder -transplantation.
Die CT wird meist mit Kontrastmittelgabe in verschiedenen Phasen durchgeführt:
- Arterielle Phase (20–30 s): optimale Darstellung arterieller Gefäße und hypervaskulärer Läsionen (z.B. HCC).
- Portalvenöse Phase (60–70 s): Beurteilung des Leberparenchym, der Pfortader und Milz
- Spätphase (3–5 min): Differenzierung von Fibrosesepten, Tumorgewebe und Narben.
In der Anfangsphase ist das CT in 25 % d.F. unauffällig. Im Verlauf finden sich dann folgende Zeichen:
- Leberarchitektur: unregelmäßige, höckrige Oberfläche, inhomogenes Parenchym, verkleinerte Leber mit Hypertrophie des linken Lappens oder Lobus caudatus.
- Regeneratknoten sind meist isodens zum restlichen Leberparenchym. Bei starker Fibrose können sie in der Spätphase leicht hypodens erscheinen. Eisenhaltige Knoten können hyperdens sein. Dysplastische Regeneratknoten zeigen eine geringe arterielle Kontrastmittelanreicherung, jedoch meist ohne Wash-out.
- Portale Hypertension: Splenomegalie, erweiterte Vena portae, Nachweis porto-systemischer Kollateralen
- Aszites: freie Flüssigkeit im Abdomen, oft begleitend.
- Tumordiagnostik: hypervaskuläre Herde in der arteriellen Phase mit Wash-out in der portalvenösen Phase sind typisch für ein HCC.
CT-Fallbeispiel
Magnetresonanztomographie
Die Magnetresonanztomographie (MRT) gilt heute als Goldstandard für die nicht-invasive, kontrastmittelgestützte Tumordiagnostik und für die detaillierte Charakterisierung von Leberläsionen. Indikationen sind:
- Früherkennung und Abklärung unklarer Lebertumoren, insbesondere HCC
- Differentialdiagnose von Regeneratknoten, Dysplasien und kleinen Tumoren (< 2 cm)
- Gefäßdiagnostik bei Verdacht auf Budd-Chiari-Syndrom oder atypische Kollateralen
- Bei Patienten mit Kontraindikationen für iodhaltiges CT-Kontrastmittel
- Funktionsdiagnostik z.B. mit Gadoxetat zur Darstellung der Hepatozytenaufnahme
Verwendete MRT-Sequenzen sind:
- T1-gewichtete Sequenzen: Beurteilung der Anatomie, Fettgehalt und nekrotischer Areale.
- T2-gewichtete Sequenzen: Darstellung von Flüssigkeiten (Aszites, Zysten), entzündlichen Prozessen.
- Diffusionsgewichtete Sequenzen (DWI): hochsensitiv für Tumornachweis.
- Kontrastmittelserien
- Arterielle Phase: Hypervaskularisation von HCC.
- Portalvenöse Phase: Abgrenzung von Tumoren und Perfusionsstörungen.
- Hepatobiliäre Phase (nach 20 min) bei leberspezifischem Kontrastmittel: Darstellung funktioneller Hepatozytenaktivität.
MRT-Befunde bei Leberzirrhose sind:
- Architektur: wie im CT.
- Fibrotische Septen sind T2w-hyperintens und T1w-hypointens. Nach Kontrastmittelgabe zeigt sich initial keine Anreicherung, in der Spätphase jedoch ein deutliches Enhancement.
- Regeneratknoten sind typischerweise iso- bis leicht hyperintens in T1 und iso- bis leicht hypointens in T2. Eisenhaltige Knoten sind stark hypointens in T1w und T2w. Im Gegensatz zu HCC zeigen sie keine arterielle Hypervaskularisation und kein Washout. In der hepatobiliären Phase können sie homogen Kontrastmittel aufnehmen. Dysplastische Knoten zeigen ein inhomogeneres Signal, meist T1w-hyperintens und T2w-hypointens und eine geringe heterogene arterielle Anreicherung ohne Washout.
- Fibrosestaging: Bestimmung der Lebersteifigkeit mittels MR-Elastographie. Werte > 5 kPa sprechen für fortgeschrittene Fibrose, > 8–12 kPa sind hochverdächtig auf Zirrhose.
- Tumoren: HCC mit charakteristischer Kontrastmittelaufnahme und Wash-out, Abgrenzung zu gutartigen Regeneratknoten. Kleine arteriell hypervaskularisierte Knoten < 1 cm entsprechen meist arterioportalen Shunts oder Regeneratknoten und sollten daher zunächst überwacht werden.
- Gefäße: Beurteilung der Vena portae und Lebervenen
- Aszites und Kollateralen: differenzierte Darstellung, insbesondere von kleinen portosystemischen Verbindungen.
Endoskopie
Die Ösophago-Gastro-Duodenoskopie (ÖGD) ist essenziell zur Erkennung und Überwachung gastroösophagealer Varizen. Ein Screening wird bei allen Patienten mit Zirrhose bei Diagnosestellung empfohlen.
Leberbiopsie
Die (meist ultraschallgestützte) Leberbiopsie ist der Goldstandard zur histologischen Sicherung und zum Fibrosestaging. Sie ist jedoch meist nur bei unklarer Diagnose oder zur Abklärung seltener Ursachen erforderlich. Eine Leberbiopsie kann auch bei unklaren raumfordernden Prozessen im Rahmen der Zirrhose indiziert sein.
Bei fortgeschrittener Leberzirrhose mit bekannter Ursache sollte eine Biopsie aufgrund der damit verbundenen Risiken vermieden werden.
Funktionsdiagnostik
Die Restfunktion der Leber kann mithilfe des LiMAx-Tests quantitativ abgeschätzt werden. Er erfasst die mikrosomale Stoffwechselleistung der Leber und ist eine Ergänzung zu etablierten klinischen Scores, ohne die Gesamtfunktion der Leber vollständig abzubilden.
Messung des hepatischen Venendruckgradienten
Der HVPG kann minimal-invasiv kathetergestützt gemessen werden. Eine klinisch signifikante portale Hypertension ist definiert als ein HVPG ≥10 mmHg. Die Messung erfolgt vor TIPS-Anlage.
Screeninguntersuchungen
Bei manifester Zirrhose sind regelmäßige Screenings erforderlich, um Komplikationen frühzeitig zu erkennen:
- HCC-Screening: Sonographie alle 6 Monate, ggf. ergänzt durch AFP-Bestimmung.
- Varizen-Screening: Initiale Endoskopie, anschließend risikoadaptiertes Intervall.
- Aszites-Überwachung: regelmäßige körperliche Untersuchung und ggf. diagnostische Punktion bei klinischer Verschlechterung.
Therapie
Die Behandlung der Leberzirrhose verfolgt drei Hauptziele:
- Therapie der Grunderkrankung, um das Fortschreiten der Fibrose zu stoppen oder - in frühen Stadien - teilweise rückgängig zu machen.
- Prävention und Behandlung von Komplikationen, die durch portale Hypertension und Leberinsuffizienz entstehen.
- Verbesserung der Prognose und Lebensqualität, bis hin zur kurativen Option der Lebertransplantation.
Ein ganzheitlicher Ansatz mit regelmäßiger Überwachung, Lebensstilmaßnahmen und medikamentöser sowie interventioneller Therapie ist erforderlich.
Basismaßnahmen
Grundlegend für alle Patienten sind allgemeine supportive Maßnahmen, die das Fortschreiten der Erkrankung verlangsamen und Komplikationen vorbeugen:
- Alkoholkarenz: Ein strikter Verzicht auf Alkohol ist unabdingbar, insbesondere bei alkoholischer Leberzirrhose, aber auch bei allen anderen Formen, da Alkohol synergistisch leberschädigend wirkt. Bereits nach 6–12 Monaten Abstinenz kann sich die Prognose deutlich verbessern.
- Ernährung: Eiweißreich, normokalorisch (1,2–1,5 g Eiweiß/kg KG/Tag), um einer Sarkopenie vorzubeugen. Keine generelle Eiweißrestriktion, außer bei akuter hepatischer Enzephalopathie. Häufige kleine Mahlzeiten, späte kohlenhydratreiche Abendmahlzeit zur Verhinderung nächtlicher Katabolismusphasen.
- Bei fortgeschrittener Zirrhose: Natriumrestriktion (max. 2 g NaCl/Tag), insbesondere bei Aszites.
- Impfungen: Schutzimpfungen gegen Hepatitis A und B, Influenza und Pneumokokken. Reduziert das Risiko schwerer Infektionen, insbesondere bei eingeschränkter Immunfunktion.
- Vermeidung hepatotoxischer Medikamente (z.B. NSAR, hochdosiertes Paracetamol, bestimmte Antibiotika). Die Einnahme vieler Medikamente (z.B. Trimipramin) ist bei Leberzirrhose kontraindiziert.
- Dosisanpassung bei renaler Dysfunktion oder portaler Hypertension
- Keine NSAR bei Aszites: Gefahr akuter Nierenverschlechterung
Ursächliche Therapie der Grunderkrankung
Die Beseitigung oder Kontrolle der Ursache kann das Fortschreiten der Zirrhose aufhalten und in frühen Stadien sogar zu einer Regression der Fibrose führen. Zum Einsatz kommen z.B.
- HCV: Direkt antiviral wirksame Substanzen (DAA) mit Heilungsraten > 95 %
- HBV: Dauertherapie mit Nukleos(t)idanaloga (z.B. Tenofovir, Entecavir)
- ALD: Alkoholkarenz, Entzugstherapie, psychosoziale Betreuung
- NAFLD: Gewichtsreduktion (≥ 10 %), mediterrane Ernährung, körperliche Aktivität. Behandlung von Begleiterkrankungen.
- Autoimmunhepatitis: Immunsuppression mit Glukokortikoiden, ggf. Azathioprin.
- Cholestatische Erkrankungen: Ursodesoxycholsäure (UDCA) bei PBC. Bei PSC aktuell keine kausale Therapie
- Hämochromatose: Aderlässe, Chelattherapie (Deferoxamin).
- Morbus Wilson: Chelattherapie (D-Penicillamin), Zinksalze.
- Alpha-1-Antitrypsin-Mangel: Substitutionstherapie, ggf. Transplantation.
Therapie der portalen Hypertension
Die Senkung des portalen Drucks ist wichtig, um Komplikationen wie Varizenblutung und Aszites vorzubeugen. Zum Einsatz kommen:
- Nicht-selektive Betablocker (NSBB): Propranolol oder Carvedilol reduzieren den portalen Druck durch Senkung des Herzzeitvolumens und Vasokonstriktion im Splanchnikusgebiet. Indiziert bei allen Patienten mit mittleren/großen Varizen oder nach erster Varizenblutung.
- Endoskopische Varizenligatur: Bei mittel- bis großkalibrigen Varizen als Primärprophylaxe bei NSBB-Unverträglichkeit oder zur Sekundärprophylaxe nach Blutung.
- TIPS (Transjugulärer intrahepatischer portosystemischer Shunt): Bei therapierefraktärem Aszites oder wiederholter Varizenblutung. Führt zu deutlicher Drucksenkung, erhöht aber das Risiko einer hepatischen Enzephalopathie.
Behandlung von Komplikationen
- Aszites: Salzrestriktion (2 g NaCl/Tag), Flüssigkeitsrestriktion nur bei Hyponatriämie (< 125 mmol/l). Diuretika (Spironolacton, ggf. mit Furosemid). Bei therapierefraktärem Aszites wiederholte Parazentese und Albumin-Substitution oder TIPS.
- Spontan bakterielle Peritonitis (SBP): Sofortige antibiotische Therapie (z.B. Cefotaxim i.v.). Albumin-Infusion zur Nierenprotektion. Sekundärprophylaxe mit Norfloxacin oder Rifaximin.
- Varizenblutung:
- Akut: Vasopressin-Analoga (Terlipressin), Antibiotika, Endoskopie mit Ligatur
- Sekundärprophylaxe: Kombination aus NSBB und endoskopischer Ligatur
- Hepatische Enzephalopathie: Laktulose oral oder rektal zur Senkung des Ammoniakspiegels. Rifaximin als Zusatz bei Rezidiven. Behandlung auslösender Faktoren (Infektionen, Elektrolytstörungen, Obstipation).
- Hepatorenales Syndrom: Vasokonstriktorische Therapie (z.B. Terlipressin) und Albumingabe. Ultima Ratio Lebertransplantation.
- Hepatopulmonales Syndrom / Portopulmonale Hypertonie: Symptomatische Therapie. Ultima Ratio Lebertransplantation.
Lebertransplantation
Die Lebertransplantation ist die einzige kurative Therapie der Zirrhose im Endstadium. Sie ist indiziert bei:
- dekompensierter Zirrhose (Child-Pugh B/C)
- Komplikationen wie therapierefraktärer Aszites, rezidivierende Varizenblutungen, HRS, HPS, ACLF
- HCC innerhalb der Mailand-Kriterien
Kriterien umfassen:
- MELD-Score ≥ 15: signalisiert signifikante Erkrankungsschwere
- Prognoseverbesserung muss das Transplantationsrisiko überwiegen
Limitierend ist die Verfügbarkeit geeigneter Spenderorgane.
Quiz
Bildquelle
- Bildquelle für Flexikon-Quiz: ©Vegan Liftz / Pexels
- Bildquelle DICOM-Viewer: Datensatz freundlicherweise zur Verfügung gestellt durch die Klinik für diagnostische und interventionelle Radiologie, St. Vinzenz Hospital Köln
Quellen
- ↑ Sørensen HT, Thulstrup AM, Mellemkjar L, et al. (2003). Long-term survival and cause-specific mortality in patients with cirrhosis of the liver: a nationwide cohort study in Denmark. Journal of Clinical Epidemiology 56 (1): 88–93 PMID: 12589875
Literatur
- Ginès et al. Liver cirrhosis. Lancet. 2021;398(10308):1359-1376. doi:10.1016/S0140-6736(21)01374-X
- Tapper und Parikh. Diagnosis and Management of Cirrhosis and Its Complications: A Review. JAMA. 2023;329(18):1589-1602. doi:10.1001/jama.2023.5997
- Ge und Runyon. Treatment of Patients with Cirrhosis. N Engl J Med. 2016;375(8):767-777. doi:10.1056/NEJMra1504367
- Schuppan und Afdhal. Liver cirrhosis. Lancet. 2008;371(9615):838-851. doi:10.1016/S0140-6736(08)60383-9
- Hegazy. Unraveling Liver Cirrhosis: Bridging Pathophysiology to Innovative Therapeutics. J Gastroenterol Hepatol. Published online August 3, 2025. doi:10.1111/jgh.70037
- Bataller und Brenner. Liver fibrosis. J Clin Invest. 2005;115(2):209-218. doi:10.1172/JCI24282