Reye-Syndrom








Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
Loslegennach dem australischen Pädiater Ralph Douglas Kenneth Reye (1912–1977)
Synonym: Reye-Morgan-Baral-Syndrom
Englisch: Reye's syndrome, white liver disease
Definition
Das Reye-Syndrom ist eine seltene, akute, potenziell lebensbedrohliche metabolische Enzephalopathie mit akuter Leberfunktionsstörung. Sie tritt typischerweise 3 bis 5 Tage nach einem Virusinfekt (v. a. des oberen Respirationstraktes oder Varizellen) bei gleichzeitiger Einnahme von Salicylaten auf. Es handelt sich überwiegend um ein pädiatrisches Krankheitsbild, Einzelfälle bei Erwachsenen sind jedoch beschrieben.
Ätiologie
Als auslösende Erreger kommen u.a. Varizella-Zoster-Viren (VZV), Influenza-A-Viren (z.B. H1N1), Influenza-B-Viren oder Epstein-Barr-Viren (EBV) in Frage.
Die Behandlung des Virusinfekts mit Salicylaten wie Acetylsalicylsäure gilt als entscheidender Risikofaktor. Seit Einführung entsprechender Warnhinweise wird ASS bei Kindern und Jugendlichen nur noch bei strenger Indikationsstellung (z. B. Kawasaki-Syndrom) eingesetzt.
Epidemiologie
Durch konsequente Vermeidung von ASS ist das Reye-Syndrom heute extrem selten. Aktuelle Registerdaten aus den USA und Europa beschreiben eine Inzidenz von weniger als 0,1 Fälle pro 1 Mio. Kinder/Jahr in Industrienationen. Seit den 2000er‑Jahren sind nur noch sporadische Einzelfälle oder kleine Fallserien beschrieben.
Betroffen sind nahezu ausschließlich Kinder unter 15 Jahren, mit einem Häufigkeitsgipfel zwischen 4 und 8 Jahren. Ein Geschlechterunterschied besteht nicht.
Die Letalität liegt in aktuellen Reviews und Leitlinien (2025) bei ca. 20 bis 30 %, abhängig vom Erkrankungsstadium bei Diagnosestellung und der Geschwindigkeit der intensivmedizinischen Therapie. Bei Überlebenden sind neurologische Residuen möglich.
Pathogenese
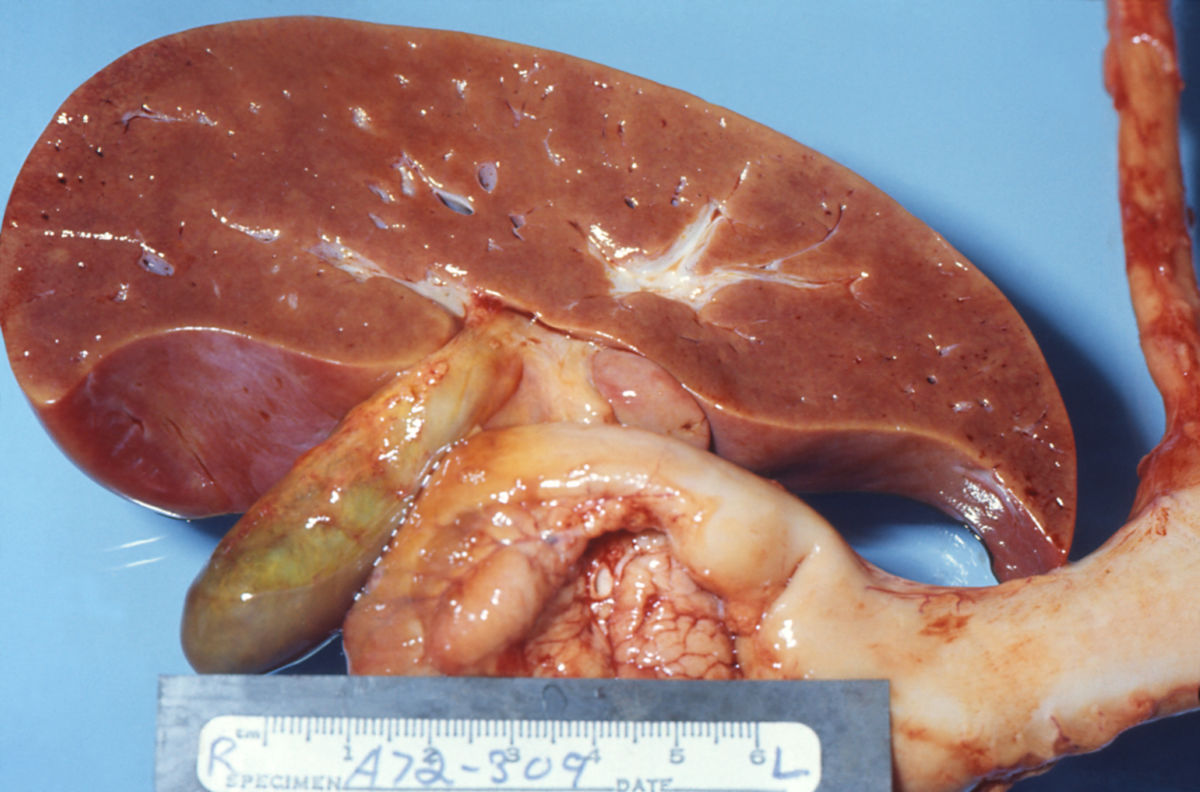
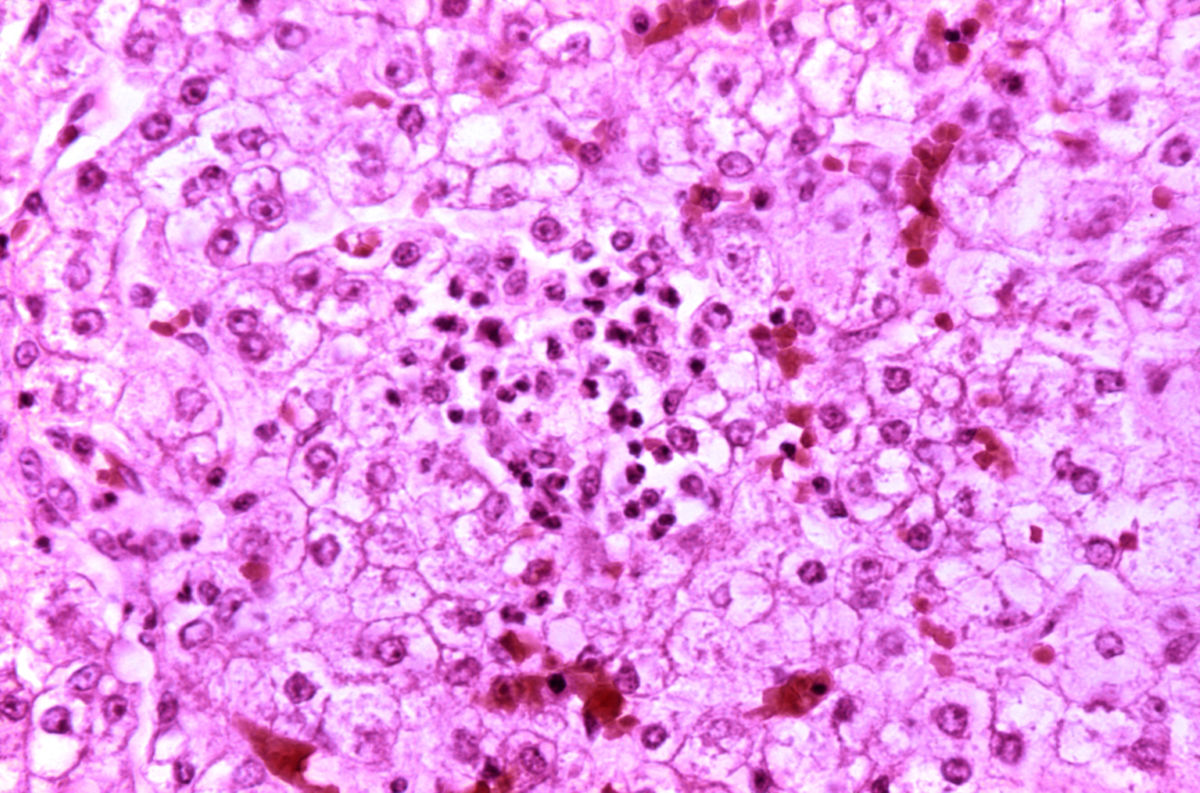
Das Reye-Syndrom beruht auf einer Fehlfunktion der Mitochondrien. In Leber, Skelettmuskel und Gehirn zeigen sie charakteristische Veränderungen mit Verlust der Cristae. Ferner findet sich in den Hepatozyten eine vermehrte Anzahl von Peroxisomen.
Der Stoffwechsel der Mitochondrien ist beeinträchtigt.
- Die Carbamoylphosphat-Synthetase, ein wichtiges Enzym im Harnstoffzyklus, weist eine verminderte Aktivität auf. Als Folge reichert sich das neurotoxische Ammoniak an.
- Die Pyruvatdehydrogenase und Enzyme der Atmungskette (Cytochrom-Oxidase) sind ebenfalls nicht voll funktionsfähig. Es findet vermehrt anaerober Stoffwechsel statt. Das dabei entstehende Laktat führt zur Azidose.
- Die Beta-Oxidation sistiert, es reichern sich langkettige Fettsäuren an.
Bei einem MCAD‑Mangel laufen ähnliche Mechanismen ab.
Das Lebergewebe unterliegt einer fettigen Degeneration (Steatosis hepatis). Ammoniak reichert sich an und führt zur Ausbildung eines Hirnödems, welches das klinische Bild einer Enzephalopathie triggert.
Symptomatik
Das Reye-Syndrom tritt in der Regel vor dem 10. Lebensjahr auf. Das Anfangsstadium des Reye-Syndroms ist gekennzeichnet durch:
- Emesis
- Somnolenz
- Lethargie
- ständiges Schreien
- Leberfunktionsstörung
Bei etwa einem Drittel der Patienten entwickelt sich in der Folge das enzephalopathische Vollbild des Reye-Syndroms:
- Hirnödem
- Hyperventilation
- Krämpfe
- Hyperreflexie mit folgender Areflexie
- Dezerebrationsstarre
- Koma
- Apnoe
Diagnose
Die Diagnose des Reye-Syndroms basiert primär auf Anamnese, typischer Klinik und Labordiagnostik.
- Anamnese (Virusinfekt, Einnahme von Salicylaten)
- Labor
- Hyperammonämie (zentrales diagnostisches Merkmal, oft > 100 bis 150 µmol/L)
- Erhöhte Transaminasen (ALAT, ASAT)
- Metabolische Azidose
- Hypoglykämie
- Erhöhte Werte für Pankreas-Amylase, Creatinkinase, Laktatdehydrogenase
- Gerinnungsparameter: INR erhöht, PTT verlängert
Darüber hinaus bestehen erhöhte Werte von Aminosäuren, freien Fettsäuren, Harnsäure und Phosphat. Angeborene Stoffwechseldefekte sollten mittels erweiterten Stoffwechselscreenings ausgeschlossen werden.
In der Bildgebung (Kranielles CT oder MRT) lässt sich ein diffuses Hirnödem nachweisen.
Differentialdiagnose
Differentialdiagnostisch sind in Betracht zu ziehen:
- Intoxikation mit Aflatoxinen, Alkaloiden oder Drogen
- Schock
- Meningitis
- Nierenversagen
- Diabetes mellitus
- angeborene Mitochondriopathien
Therapie
Die Therapie ist intensivmedizinisch, symptomatisch und supportiv. Kausale Therapiekonzepte sind nicht vorhanden.
Der Patient wird intubiert und sediert. Der Hirndruck muss invasiv überwacht werden, zur Verminderung können osmotische Diuretika wie z.B. Mannitol dienen. Die Hyperammoniämie kann mittels Peritonealdialyse behandelt werden.
Das Vollbild des Reye-Syndroms führt in über 75% der Fälle zum Tod. Eine früh einsetzende Therapie im Anfangsstadium der Erkrankung kann die Mortalität deutlich vermindern.
Quellen
- Gelbe Liste: Reye Syndrom (abgerufen am 5.12.25)
- Schrör, Aspirin and Reye syndrome: a review of the evidence, Paediatr Drugs, 2007