Hereditäre Koproporphyrie




Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenEnglisch: hereditary coproporphyria
Definition
Die hereditäre Koproporphyrie, kurz HCP, ist eine seltene hereditäre Erkrankung aus der Gruppe der akuten hepatischen Porphyrien. Sie ist durch das attackenartige Auftreten neuroviszeraler Symptome und teils Photosensibilität charakterisiert.
Epidemiologie
Die Prävalenz in Europa wird auf etwa 1 zu 1.000.000 geschätzt. Es sind vorwiegend Frauen betroffen.
Ätiopathogenese
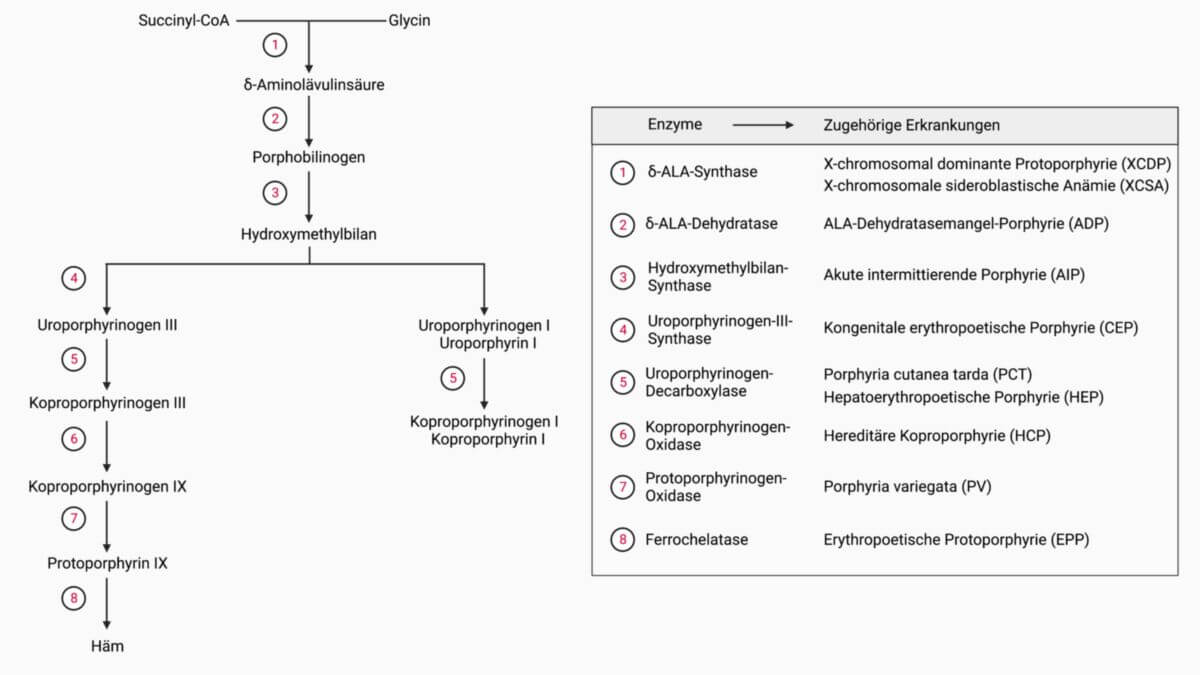
Ursächlich für die Erkrankung ist ein Defekt der Koproporphyrinogen-Oxidase (CPO). Die Mutation befindet sich im CPOX-Gen am Genlokus 3q12. Die Vererbung erfolgt nach autosomal-dominantem Muster mit geringer Penetranz. Die CPO ist ein Enzym der Hämsynthese. Durch die verringerte Aktivität kommt es bei Stimulation der Hämsynthese zur Akkumulation von Porphyrinen und Porphyrin-Vorläufer-Molekülen in der Leber.
Folgende Faktoren können eine Attacke auslösen und sollten entsprechend gemieden werden:
- porphyrinogene Medikamente
- Alkohol
- Infektionen
- Fasten
- Stress
- hormonelle Änderungen (z.B. Menstruationszyklus, Kontrazeptiva)
Symptome
Die Symptome treten in Attacken auf. Im Vordergrund stehen dabei starke viszerale Schmerzen, die sich über Tage entwickeln können. Hinzu kommen oft Übelkeit, Erbrechen, Obstipation und Lumbago.
Auch die Ausbildung neurologischer Symptome ist möglich. Wird die Attacke nicht behandelt, kann sich z.B. eine motorische Neuropathie entwickeln. Diese beginnt typischerweise proximal in Armen und Beinen und breitet sich langsam nach distal aus. Zusätzliche neurologische Symptome sind Myalgien, Paresen oder epileptische Anfälle.
Mögliche psychiatrische Symptome sind Reizbarkeit, depressive Symptome und das Auftreten von Ängsten. In seltenen Fällen sind visuelle und akustische Halluzinationen sowie Verwirrtheitszustände möglich.
Es kommt teils zu Tachykardien und einer behandlungswürdigen Hyponatriämie.
In seltenen Fällen entwickeln sich potentiell letale kardiale Arrhythmien oder eine Zwerchfellparese.
Etwa 20 bis 30 % der Betroffenen haben infolge der entstehenden erhöhten Photosensibilität auch kutane Läsionen. Die entstehenden Bullae hinterlassen teils hyperpigmentierte Narben.
Diagnostik
Im Blut kann eine Erhöhung der Porphyrine (δ-Aminolävulinsäure und Porphobilinogen) nachgewiesen werden. Im Urin sind Uroporphyrine und Coproporphyrine erhöht. Es fällt teils ein gelbroter oder bräunlicher Urin auf.
Der molekulargenetische Nachweis einer heterozygoten pathogenen Mutation im CPOX-Gen beweist die Erkrankung.
Im Verlauf sollte bei erhöhtem δ-Aminolävulinsäure-Level und bei einem Alter von über 60 Jahren die Leber- und Nierenfunktion jährlich überprüft werden. Zudem ist ein Screening auf Leberfibrose sowie auf das Vorliegen eines hepatozellulären Karzinoms (ab 60 Jahre) empfohlen.[1]
Differentialdiagnosen
Differentialdiagnostisch kommen die akute intermittierende Porphyrie und die Porphyria variegata in Betracht.
Therapie
In der akuten Attacke kommen Infusionen mit Hämatin zum Einsatz. Zudem kann eine Infusion von Kohlenhydraten sinnvoll sein. Eine Hyponatriämie wird ausgeglichen.
Auslösende Faktoren (s.o.) müssen so schnell wie möglich eliminiert werden. Es erfolgt zudem eine symptomatische Behandlung der Schmerzen, Übelkeit und anderer auftretender Symptome. Hierbei müssen porphyrinogene Medikamente vermieden werden.
Bei Frauen kann eine Ovulationshemmung mit GnRH-Agonisten erwogen werden, wenn die Attacken an den Menstruationszyklus gebunden sind.[1]
In einigen Fällen ist eine Lebertransplantation nötig, wenn trotz Behandlung Attacken und bleibende neurologische Schäden auftreten. Hautläsionen können durch ein konsequenten Schutz vor Sonneneinstrahlung vermieden werden.
Prognose
Die Attacken treten seltener auf als bei der akuten intermittierenden Porphyrie. Durch konsequente Vermeidung auslösender Faktoren kann eine Progression der Erkrankung meist verhindert werden.
Dennoch kommt es teils zur Entstehung bleibender neurologischer Störungen. Auch letale Ausgänge sind (selten) möglich, insbesondere bei Herzrhythmusstörungen und Atemlähmung.
Literatur
- orpha.net – Hereditary coproporphyria, abgerufen am 16.06.2023
Quellen
- ↑ 1,0 1,1 Wang et al., Hereditary Coproporphyria. GeneReviews, 2012