Dup15q-Syndrom




Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenSynonyme: Mikroduplikationssyndrom 15q11q13, 15q11q13-Duplikationssyndrom, Dup(15)(q11q13), Trisomie 15q11q13
Englisch: Dup15q syndrome, 15q11.2-q13.1 duplication syndrome, dup15q syndrome
Definition
Das Dup15q-Syndrom, kurz für Chromosom 15q11.2-q13.1 Duplikationssyndrom, ist eine seltene genetische Erkrankung, die unter anderem durch Entwicklungsverzögerungen, Muskelhypotonien und Autismus-Spektrum-Störungen gekennzeichnet ist. Ursächlich sind Duplikationen in der sogenannten Prader-Willi/Angelman-kritischen Region (PWACR).
Epidemiologie
Die genaue Prävalenz des Dup15q-Syndroms ist derzeit (2026) noch nicht bekannt. Sie wird auf 1:5.000 bis 1:20.000 geschätzt.
Ätiologie
Das Dup15q-Syndrom wird durch chromosomale Anomalien verursacht, wodurch mindestens eine zusätzliche Kopie der Region 15q11.2-q13.1 von Chromosom 15 vorliegt. Die Erkrankung tritt jedoch nur auf, wenn diese Anomalie von mütterlicher Seite vererbt wird.
Genetik
Die betroffene Region auf Chromosom 15 wird als Prader-Willi/Angelman-kritische Region (PWACR) bezeichnet, da Deletionen des Abschnitts das Prader-Willi-Syndrom und Angelman-Syndrom auslösen. Zu den Proteinen, die in diesem Abschnitt genetisch kodiert sind, zählen u.a. die Flippase ATP10A, die Ubiquitinligase UBE3A sowie verschiedene Untereinheiten des GABA-A-Rezeptors. Die Duplikation einzelner Gene am Genlokus führt nicht zu einem Dup15q-Syndrom.
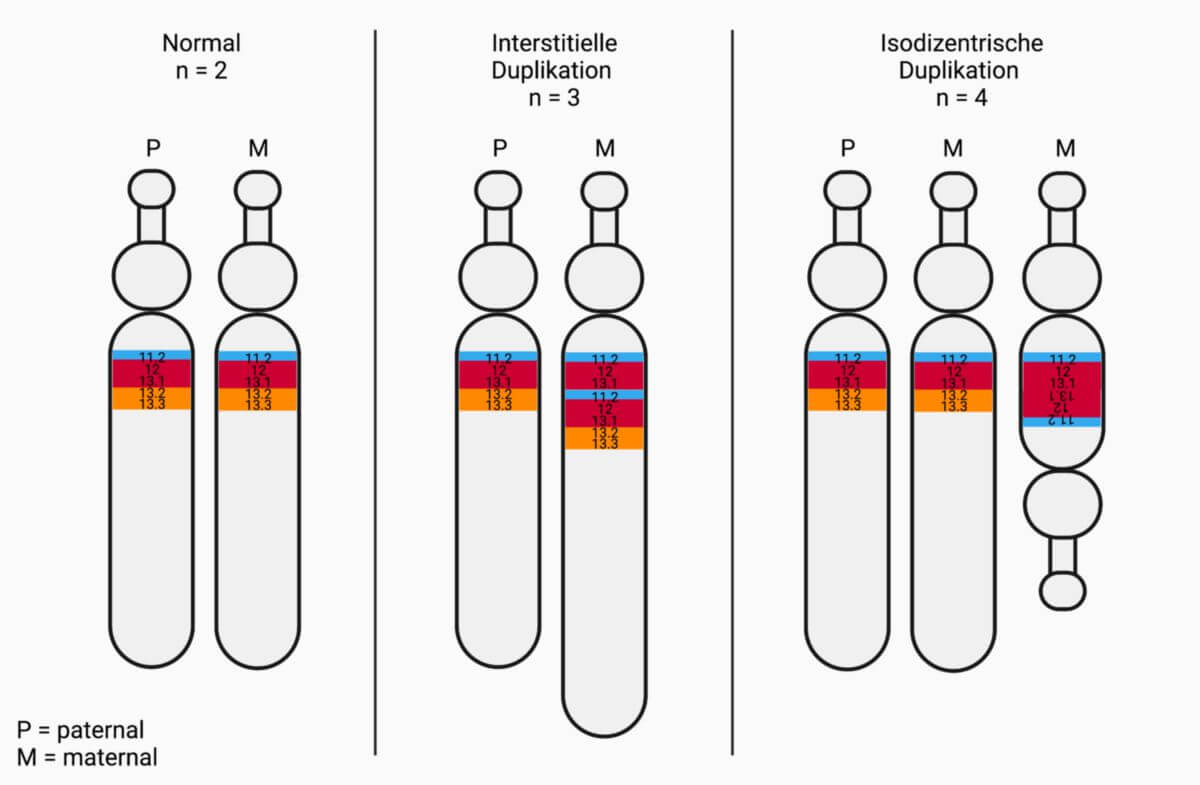
Die Chromosomenaberration, die beim Dup15q-Syndrom am häufigsten auftritt (~ 80 % d.F.), ist ein isodizentrisches Chromosom 15, kurz idic(15). Dieses liegt als zusätzliches Chromosom neben den beiden normalen Kopien vor und enthält zwei gespiegelte PWACRs, die sich zwischen zwei Centromeren befinden. Individuen haben dementsprechend vier Kopien des Segments.
Bei ~ 20 % der Betroffenen befindet sich die Duplikation innerhalb des langen Arms (q-Arm) auf einem der beiden Chromosomen. Diese Form wird als interstitielle Duplikation, kurz int dup(15), bezeichnet. Hierbei handelt es sich meist um eine Neumutation. Im Vergleich zu idic(15) liegen nur drei Kopien der PWACR vor, entsprechend weisen die Patienten meist mildere Verläufe auf.
Sehr selten treten andere Formen der Aberrationen auf, z.B. interstitielle Triplikationen.
Imprinting
In der PWACR befinden sich einige Gene, die durch genomisches Imprinting reguliert werden. Durch epigenetische Modifikationen (z.B. DNA-Methylierung) sind bestimmte Gene nur auf dem mütterlichen, andere nur auf dem väterlichen Chromosom aktiv.
Das Dup15q-Syndrom wird nur durch Aberrationen des mütterlichen Chromosoms ausgelöst. Dies lässt darauf schließen, dass primär solche Gene für die Erkrankung verantwortlich sind, die nur vom mütterlichen Chromosom exprimiert werden (z.B. ATP10C und UBE3A).
Klinik
Das Dup15q-Syndrom ist durch unterschiedliche Symptome charakterisiert. Die Erkrankung ist unter anderem durch Entwicklungsverzögerungen und Autismus-Spektrum-Störungen gekennzeichnet. Der Schweregrad der Entwicklungsstörungen kann interindividuell variieren. Insbesondere die Sprachentwicklung ist im Rahmen des Dup15q-Syndroms häufig verzögert.
Säuglinge und Neugeborene weisen typischerweise eine muskuläre Hypotonie auf, die mit Problemen bei der Nahrungsaufnahme einhergeht. Viele betroffene Kinder zeigen eine verzögerte Entwicklung der Grob- und Feinmotorik. Trotz des erniedrigten Muskeltonus können die meisten betroffenen Kinder im Alter zwischen zwei und drei Jahren eigenständig gehen. Der Gang ist in der Regel breitbeinig und ataktisch.
Bei über 50 % der Patienten mit dem Dup15q-Syndrom liegt eine Epilepsie vor. Die ersten Krampfanfälle manifestieren sich in der Regel zwischen dem sechsten Lebensmonat und neunten Lebensjahr. Beim Dup15q-Syndrom handelt es sich um eine der häufigsten Ursachen für das West-Syndrom. Einige Patienten weisen ein Lennox-Gastaut-Syndrom auf.
Patienten mit dem Dup15q-Syndrom können verschiedene Gesichtsdysmorphien aufweisen, diese sind meist nur subtil und werden häufig übersehen. Beispiele für Gesichsmerkmale sind:
- Makrozephalie
- abgeflachter Nasenrücken und gebogene Nasenspitze
- langes Philtrum
- antevertierte Nasenlöcher
- nach unten gerichtete Lidspalten
- Epikanthus
- Mikrognathie
- tief angesetzte Ohren
- flaches Hinterhaupt
- niedrige Stirn
Weitere Symptome, die im Rahmen des Dup15q-Syndroms auftreten können, sind beispielsweise:
Diagnostik
Die Verdachtsdiagnose ergibt sich durch das klinische Erscheinungsbild. Gesichert wird die Diagnose durch molekulargenetische Untersuchungen wie beispielsweise eine Fluoreszenz-in-situ-Hybridisierung oder ein DNA-Microarray. Zudem wird den betroffenen Familien eine genetische Beratung angeboten.
Therapie
Die Behandlung erfolgt in der Regel durch ein multidisziplinäres Team und umfasst unter anderem Ergo- und Physiotherapie sowie Verhaltenstherapien. Darüber hinaus benötigen die Patienten eine antikonvulsive Therapie. Die Patienten müssen regelmäßig untersucht und die motorische und sprachliche Entwicklung beurteilt werden.
Literatur
- Orphanet - Dup15q-Syndrom, abgerufen am 15.08.2022
- Rarediseases - Dup15q Syndrome, abgerufen am 15.08.2022
- Dup15q, abgerufen am 15.08.2022