Retinoblastom









Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenEnglisch: retinoblastoma
Definition
Das Retinoblastom, kurz RB, ist eine seltene maligne Neoplasie der Netzhaut. Es tritt vor allem im Kindesalter auf und zählt zu den dysontogenetischen Tumoren.
Epidemiologie
Im Kindesalter ist das Retinoblastom der am häufigsten vorkommende Augentumor und macht etwa 2 % aller Krebserkrankungen in dieser Altersgruppe aus. Das Retinoblastom manifestiert sich in den meisten Fällen vor dem 3. Lebensjahr, selten auch später. In wenigen Fällen tritt es sogar erst im Jugendalter auf.
Ätiologie
Das Retinoblastom ist eine genetisch bedingte Neoplasie. Voraussetzung für die Manifestation ist die Inaktivierung beider Allele des Retinoblastom-Gens (RB1) (Knudson-Hypothese). Hierbei handelt es sich um ein Tumorsuppressorgen, das auf Chromosom 13 am Genlokus 13q14 kodiert ist. Die Inaktivierung erfolgt durch Deletionen und andere Mutationen und kann bereits in den Keimzellen oder erst in den somatischen Zellen der Retina stattfinden.
Einteilung
Retinoblastome lassen sich nach ihrer Ätiologie in zwei Formen unterteilen:
- hereditäres/familiäres Retinoblastom (5 bis 10 % der Fälle): hierbei ist das erste Allel bereits durch eine Keimbahnmutation betroffen, das zweite wird durch ein spontanes Mutationsereignis inaktiviert
- sporadisches Retinoblastom (90 bis 95 % der Fälle): beide Allele werden durch spontane Mutationsereignisse in den somatischen Zellen inaktiviert
Humangenetische Aspekte
Mutationen in Keimzellen führen dazu, dass alle retinalen Zellen ein erhöhtes Entartungsrisiko aufweisen. In diesem Fall treten Retinoblastome meistens multilokulär (an mehreren Stellen der Netzhaut) bzw. beidseitig auf. Keimbahnmutation werden autosomal-dominant vererbt, jedoch mit unvollständiger Penetranz.
Bei somatischen Mutationen bestimmter Zellen eines Netzhautareals kommt es häufiger zu einem unilokulären, einseitigen Retinoblastom (55 bis 60 % aller Patienten). Nur ca. 35 % aller Patienten weisen hier eine beidseitige Form auf.
Für die humangenetische Beratung ergeben sich aus der Art des Retinoblastoms und der Familienanamnese wertvolle Hinweise:
- Bei unauffälliger Familienanamnese und unilokulärem Retinoblastom ist das Erkrankungsrisiko für weitere Kinder desselben Paares relativ gering.
- Bei multilokulärem Befall und auffälliger Familienanamnese ("Der als Kind gestorbene Bruder von Opa hatte auf alten Fotos immer ein weißes Auge") ist das Erkrankungsrisiko relativ hoch, da eine autosomal-dominant erbliche Keimbahnmutation anzunehmen ist.
Eine detaillierte und definitive Beratung sollte stets zusammen mit einem Augenarzt und einem Humangenetiker erfolgen. In diesem Rahmen sind zudem weiterführende molekulargenetische Untersuchungen möglich.
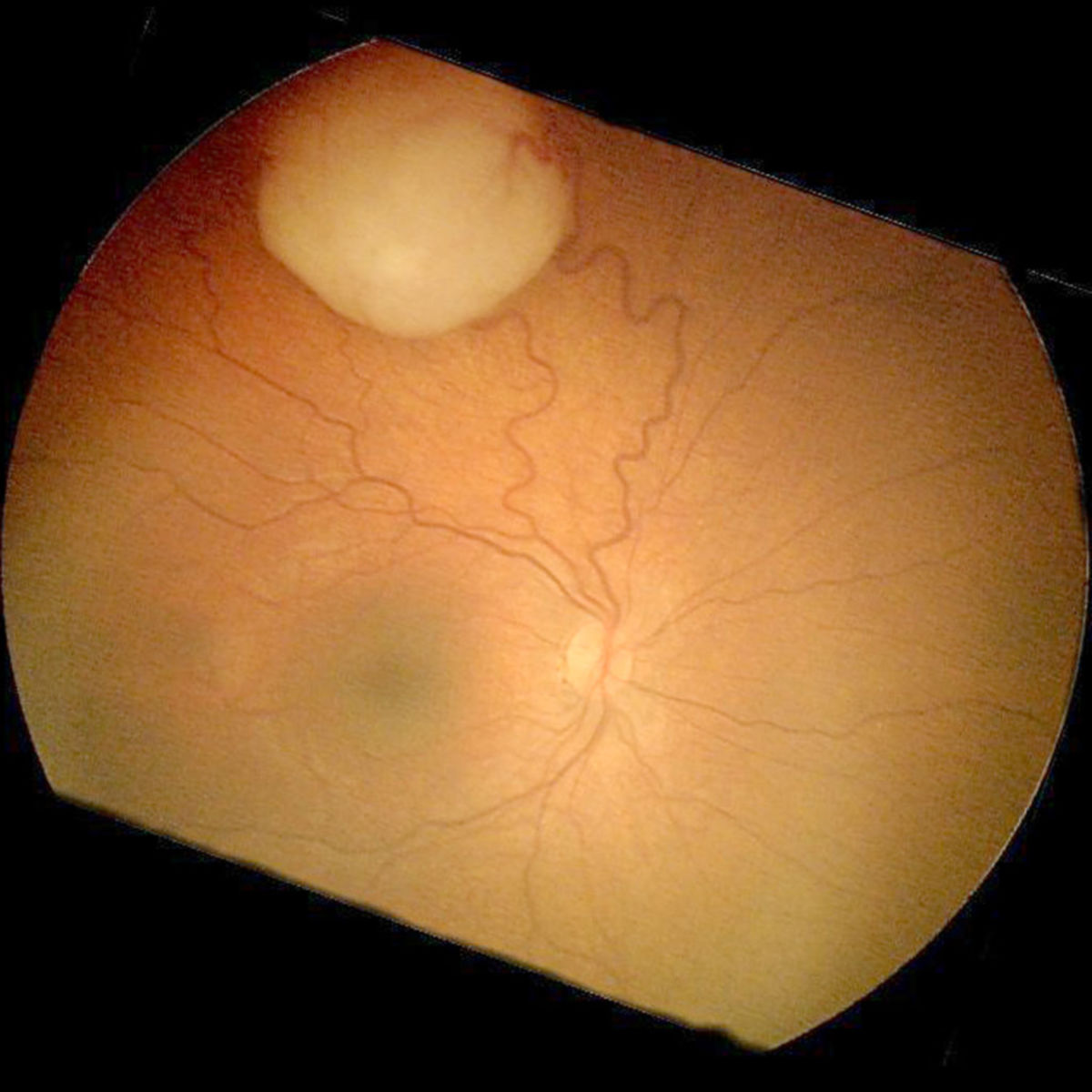
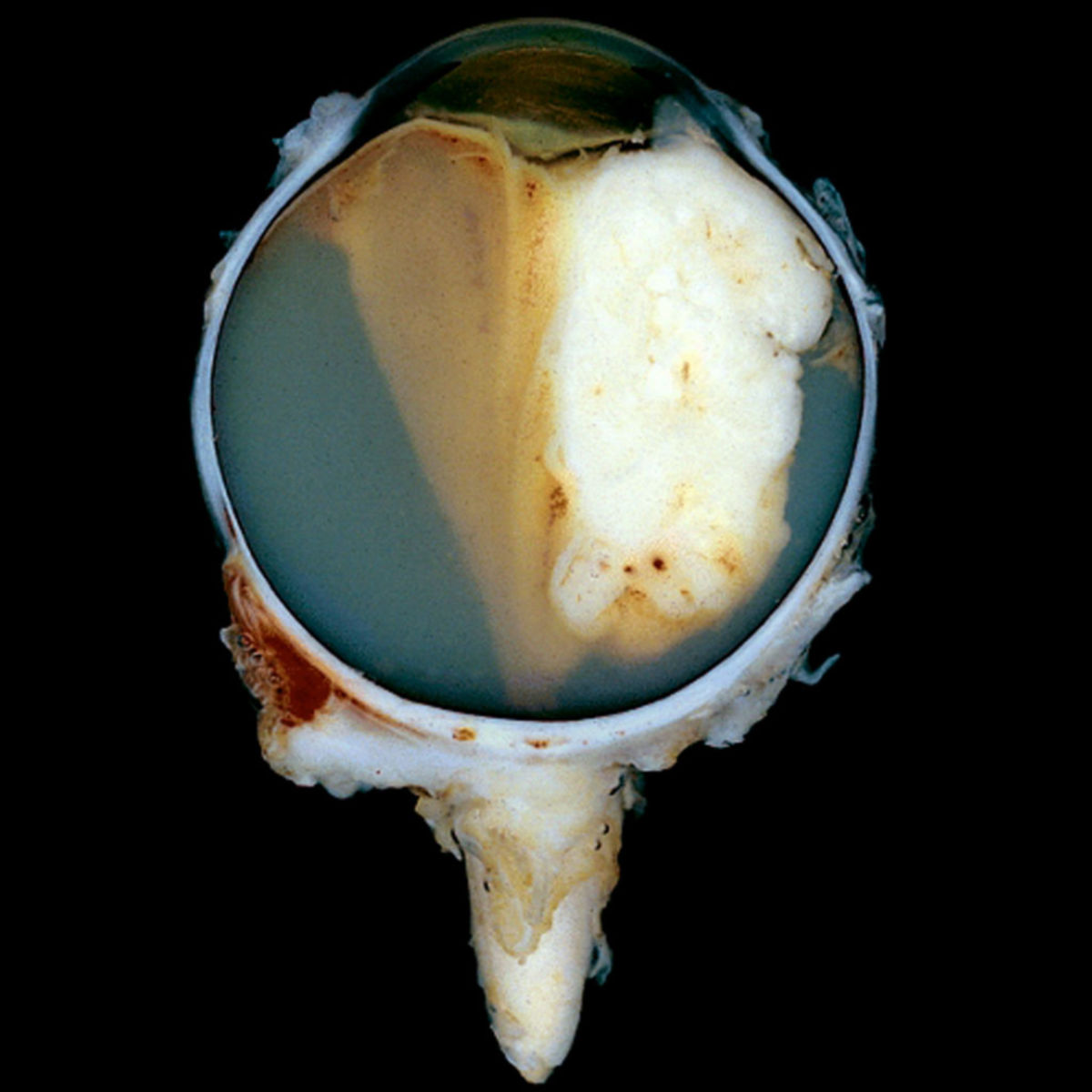
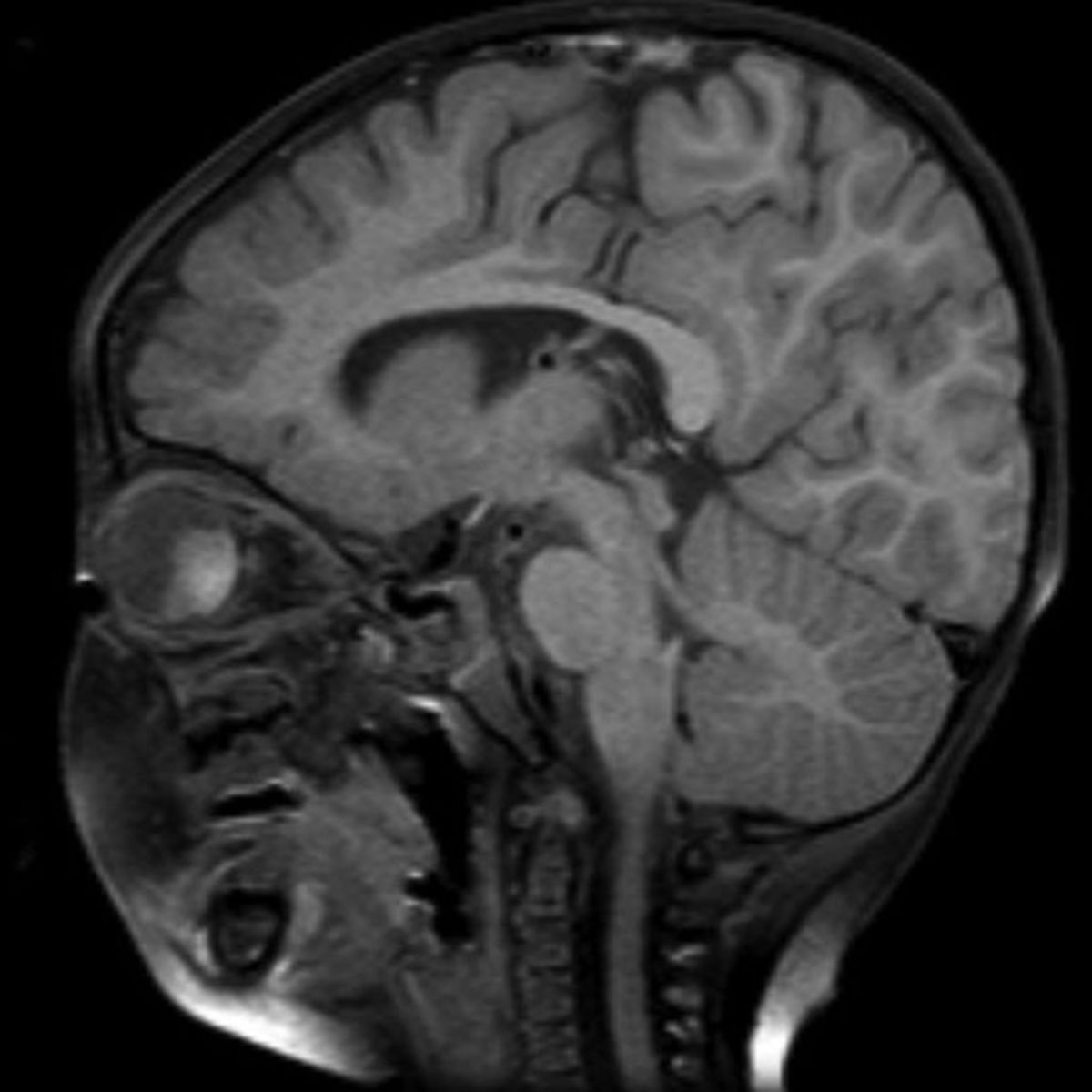
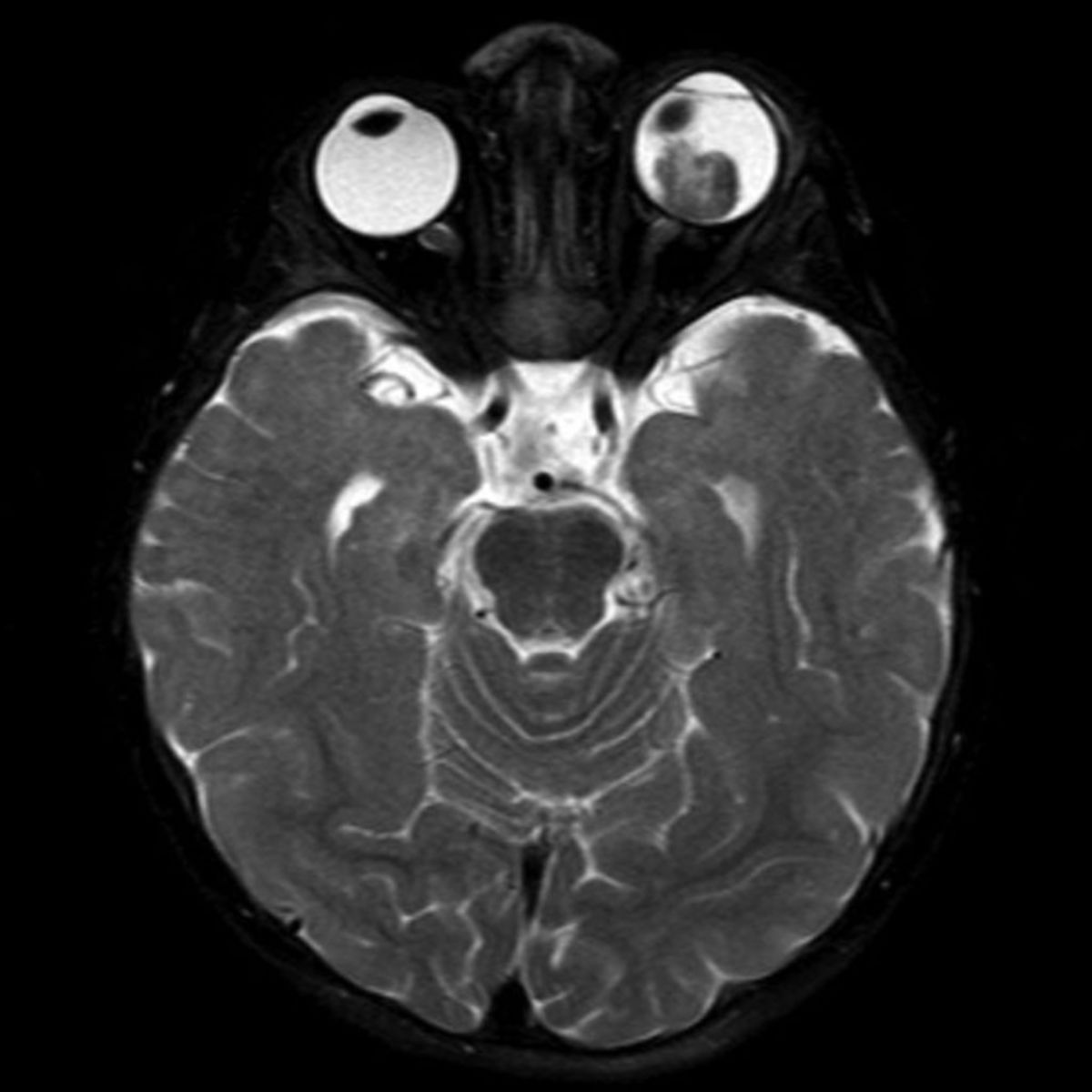
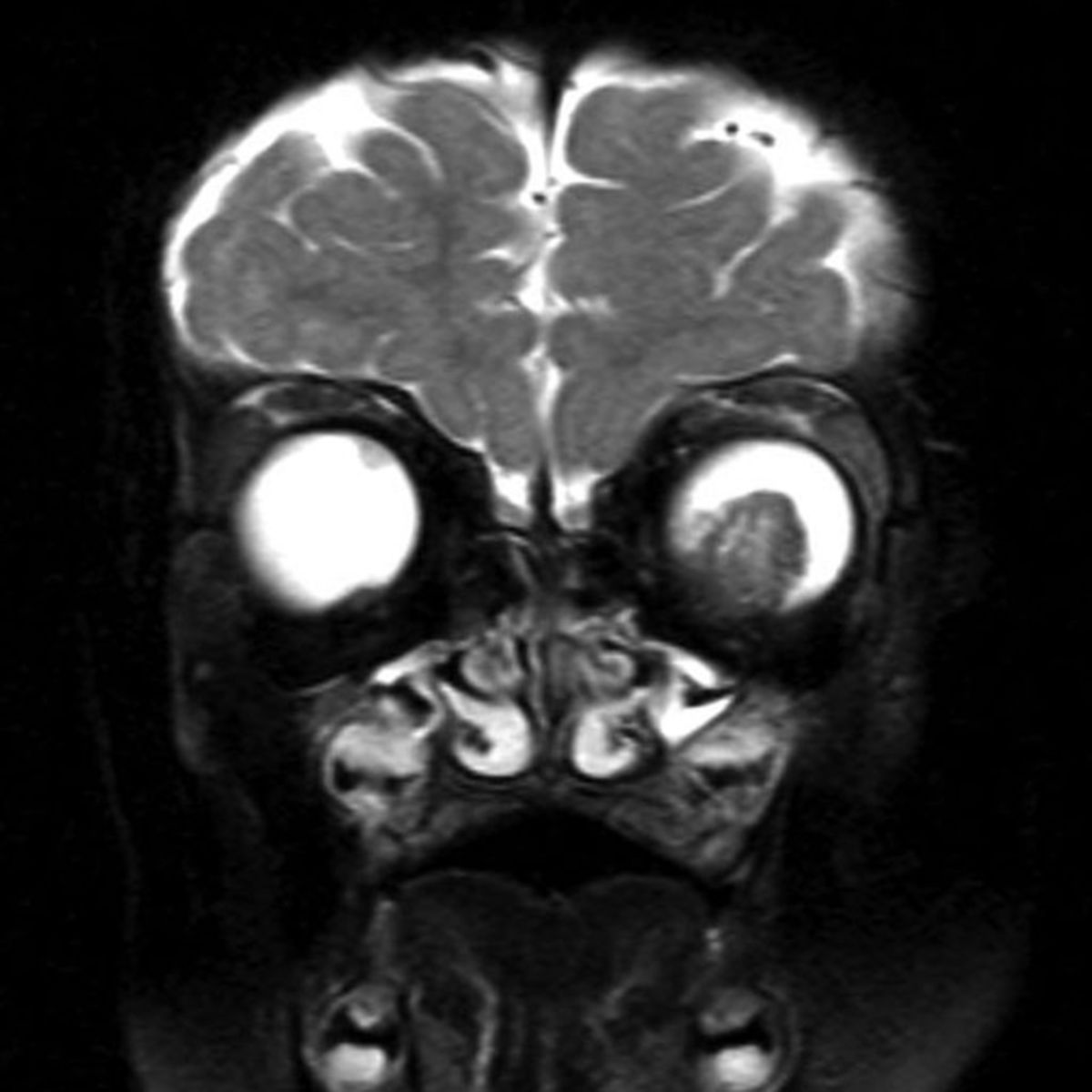
Symptomatik
In der überwiegenden Zahl der Fälle stellen sich die Patienten mit einer Leukokorie (weiße Pupille, Katzenauge) oder einem durch Einwachsen in zentrale Bereiche der Netzhaut entstehenden Strabismus vor. Weitere, seltenere Symptome können Entzündungen, Visusverlust, erhöhter Augeninnendruck (Glaukom) oder ein Exophthalmus sein.
Ein Retinoblastom entsteht in seltenen Fällen auch im Corpus pineale (trilaterales Retinoblastom) und führt dann zu einer vorrangig neurologischen Symptomatik. Diese wird unter anderem durch verstärkten Befall der Hirnhäute verursacht.
Diagnose
Die Diagnose erfolgt in der Regel durch einen Augenarzt. Vor allem einseitige Retinoblastome werden häufig erst spät erkannt, sodass das betroffene Auge bereits erblindet sein kann.
Die Diagnostik umfasst:
- Fundoskopie (Betrachtung des Augenhintergrundes)
- MRT, CT und Sonographie (zum Nachweis von Kalzifikationen und Beurteilung der Ausbreitung)
- molekulargenetische Untersuchung
- ggf. Metastasensuche
Therapie
Kleine Tumoren werden lokal behandelt. Zum Einsatz kommen hierbei Laser, Brachytherapie oder temperaturabhängige Verfahren wie Kryotherapie und Thermochemotherapie. Zur Reduzierung der Tumormasse wird vor der Therapie teils eine systemische Chemotherapie angewandt (u.a. Vincristin und Carboplatin).
Größere Tumoren erfordern die Enukleation des Auges mit einer ausgiebigen Resektion des Nervus opticus. Zusätzlich ist teils eine adjuvante Chemotherapie oder Bestrahlung notwendig. Bei Entfernung des Augapfels wird zudem im weiteren Verlauf eine Prothese eingesetzt.
Erfolgreich behandelte Patienten sollten regelmäßig nachuntersucht und beim Auftreten verdächtiger Befunde schnellstmöglich erneut therapiert werden. Bei einem Teil der Patienten entwickeln sich sowohl Retinoblastome des zweiten Auges oder des Corpus pineale, als auch andere maligne Neoplasien wie Sarkome (z.B. Osteosarkom).
Prognose
Die Mortalität des Retinoblastoms liegt bei adäquater Therapie und früher Diagnose bei unter 5 %. Zu beachten ist jedoch das erhöhte Risiko für das Auftreten weiterer Malignome bei der hereditären Form. Ebenfalls können vorhandene Metastasen die Prognose massiv verschlechtern. Unbehandelt liegt die Sterberate bei über 99 %.
Literatur
- Pschyrembel - Retinoblastom, abgerufen am 20.06.2022
- MSD Manual - Retinoblastom, abgerufen am 20.06.2022
- Orpha.net - Retinoblastom, abgerufen am 20.06.2022
- Kinderkrebsinfo - Retinoblastom, abgerufen am 20.06.2022
- KinderAugenKrebsStiftung - Unilaterales Retinoblastom, abgerufen am 20.06.2022
- KinderAugenKrebsStiftung - Trilaterales Retinoblastom, abgerufen am 20.06.2022
- Othman. Retinoblastoma major review with updates on Middle East management protocols. Saudi Journal of Ophthalmology. 26(2):163-175. 2012