Kaposi-Sarkom









Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
Loslegennach dem österreichisch-ungarischen Hautarzt Moritz Kaposi (1837-1902)
Synonyme: Sarcoma idiopathicum multiplex haemorrhagicum, Morbus Kaposi
Definition
Das Kaposi-Sarkom, kurz KS, ist eine vermutlich durch das Humane Herpesvirus 8 (HHV-8) induzierte Neoplasie lymphatischer Endothelzellen. Es kann Haut- und Schleimhäute sowie innere Organe betreffen. Das Kaposi-Sarkom ist ein niedrig-gradiger, infiltrativ und lokal-aggressiver, selten metastasierender Tumor.
ICD11-Codes
- 1C60.30 - Kaposi-Sarkom assoziiert mit HIV-Erkrankung assoziiert mit Malaria
- 1C61.30 - Kaposi-Sarkom assoziiert mit HIV-Erkrankung assoziiert mit Tuberkulose
- 2B57 - Primäres Kaposi-Sarkom
- 2B57.0 - Kaposi-Sarkom der Lunge
- 2B57.1 - Kaposi-Sarkom der Haut
- 2B57.2 - Kaposi-Sarkom des Gastrointestinaltrakts
- 2B57.Y - Kaposi-Sarkom in anderer Region
- 2B57.Z - Kaposi-Sarkom, nicht näher bezeichnet
Einteilung
Man unterscheidet fünf Formen:
- sporadisches, klassisches Kaposi-Sarkom
- Kaposi-Sarkom bei iatrogener Immunsuppression
- endemisches (afrikanisches) Kaposi-Sarkom
- epidemisches, HIV-assoziiertes und Immunrekonstitutionssyndrom (IRIS)-assoziiertes Kaposi-Sarkom
- Kaposi-Sarkom bei HIV-negativen Männern, die Sex mit Männern haben (MSM)
| Form | Klinik | Risikofaktoren | Verlauf |
|---|---|---|---|
| Klassisches KS |
|
|
|
| Kaposi-Sarkom iatrogener Immunsuppression |
|
|
|
| Endemisches KS |
|
|
|
| HIV- und IRIS-assoziiertes KS |
|
|
|
| KS bei MSM ohne HIV-Infektion |
|
|
|
Epidemiologie
Entsprechend der Pathogenese korreliert die Prävalenz des Kaposi-Sarkoms mit der Verbreitung von HHV-8-Infektionen. Die HHV-8-Seroprävalenz erreicht über 40 % in der Subsahara-Region, 10 bis 30 % im Mittelmeerraum und < 5 % in Nordeuropa. Die Inzidenz in der Allgemeinbevölkerung liegt bei ca. 1,5 Fällen pro 100.000 Personen pro Jahr. Bei einer HIV-Infektion liegt die Inzidenz bei ca. 482 pro 100.000 Personen pro Jahr, wobei die Inzidenz unter effektiver antiretroviraler Therapie auf 181 pro 100.000 Personen pro Jahr sinkt.
Klassisches KS
Das klassische KS tritt bei Männern 2- bis 17-mal häufiger auf. Betroffen sind insbesondere Personen osteuropäischer und mediterraner Herkunft. Die Erstmanfestation erfolgt typischerweise nach dem 60. Lebensjahr. Risikofaktoren sind:
- vorangegangene HHV-8-Infektion
- erhöhtes Alter
Die Mortalität ist bei Patienten mit klassischem KS im Vergleich zur Normalbevölkerung nicht wesentlich gesteigert.
Endemisches KS
Das endemische KS findet sich in Äquatorialafrika und ist nicht mit einer HIV-Infektion assoziiert. Es ist vor allem bei Kindern und adoleszenten Männern verbreitet.
KS bei iatrogener Immunsuppression
Bei langfristiger Immunsuppression können iatrogene KS entstehen. Nach Absetzen oder Umstellung der Medikation kann sich die Erkrankung wieder zurückbilden. Kaposi-Sarkome machen 5 % der Malignome nach Organtransplantation aus. Eine Organtransplantion erhöht das Risiko um das 50 bis 500fache im Vergleich zur Normalbevölkerung.
Epidemisches, HIV-assoziierte KS
Das epidemische, HIV-assoziierte KS ist die häufigste AIDS-definierende Neoplasie und der häufigste Subtyp des KS. Der Verlauf ist sehr variabel. Nach Einleitung einer antiretroviralen Therapie bilden sich HIV-assoziierte KS häufig ohne zusätzliche Behandlung zurück. Bei Immunrekonstitutionssyndrom (IRIS) wurden schwere und neuaufgetretene Kaposi-Sarkome beschrieben. Die Mortalität ist assoziiert mit der CD4-Zellzahl.
KS bei MSM ohne HIV-Infektion
Kaposi-Sarkome treten zunehmend häufiger bei jüngeren HIV-negativen MSM aus geographischen Regionen mit einer niedrigen HHV-8-Seroprävalenz (z.B. Frankreich, England oder Deutschland) auf. Der Verlauf ähnelt dem klassischen KS. CD4-Zellzahl und CD4/CD8-Ratio korrelieren mit der Krankheitsschwere.
Ätiopathogenese
Die genaue Ursache des Kaposi-Sarkoms ist derzeit (2022) unklar. Ob es sich um eine maligne Neoplasie oder um eine angioproliferative Erkrankung handelt, ist umstritten. Eine HHV-8-Infektion führt nicht zwangsläufig zur Manifestation einer HHV-8-assoziierten Erkrankung, jedoch sind alle 5 Subtypen mit einer HHV-8-Infektion assoziiert. In über 95 % aller Fälle lässt sich die Virus-DNA nachweisen. Das Virus infiziert lymphatische Endothelzellen, führt zur Störung ihrer mesenchymalen Differenzierung, induziert die Zytokinexpression und fördert eine abnormale Neoangiogenese.
HHV-8 wird insbesondere über Speichel, aber auch sexuell, vertikal und über Blutprodukte übertragen. Weitere mit HHV-8-assoziierte Malignome sind der multizentrische Morbus Castleman (MCD), primäre Effusionslymphome, intravaskuläre großzellige B-Zell-Lymphome (IVBZL), Angiosarkome, und inflammatorische myofibroblastische Tumore.
Das Auftreten und die Progression von KS wird weiterhin durch Immundefekte, Immunsuppression und/oder niedrige CD4-Zellzahlen begünstigt. Außerdem wird eine genetische Prädisposition vermutet.
Klinik
Klassisches KS
Das klassische KS ist charakterisiert durch uni- oder bilateral auftretende, rote oder schwarzbraune Maculae, die oft mit Ödemen assoziiert sind. Typischerweise ist die untere Extremität beteiligt. Die Flecken entwickeln sich im Verlauf zu rotbraunen Plaques und knotigen Tumoren. Verschiedene Stadien können dabei zeitgleich nebeneinander auftreten. Die einzelnen Tumoren können über Jahre unverändert bleiben oder sich rasch ausbreiten und an Zahl und Größe zunehmen. Lymphabflussstörungen mit deutlichen Schwellungen ganzer Extremitäten, des Skrotums oder des Gesichts sind möglich, ebenso Einblutungen, zentrale Nekrosen und Ulzerationen. Auch ausgeprägt hyperkeratotische bzw. verruköse Formen kommen vor.
Endemischer KS
Beim endemischen KS werden vier klinische Typen unterschieden:
- benigne Verlaufsform: langsam und wenig aggressiv fortschreitende noduläre Läsionen der Haut, ähnlich des klassischen KS. Insbesondere bei Männern mittleren Alters.
- lokal-aggressive Verlausform: kutane Läsionen mit aggressiver Infiltration in Weichgewebe und Knochen. Meist letaler Verlauf innerhalb 5 bis 7 Jahren.
- diffus-aggressive Verlaufsform: diffuser mukokutaner und viszeraler Befall. Ungünstige Prognose.
- fulminante Verlaufsform: Lymphadenopathie und Befall viszeraler Organe; selten Hautbefall. Fulminant-aggressiver Verlauf. Insbesondere bei Kleinkindern.
Epidemisches, HIV-assoziierte KS
Das epidemische, HIV-assoziierte KS ähnelt dem klassischen Typ, weist jedoch unterschiedliche Prädilektionsstellen auf: Das initiale Tumorgeschehen manifestiert sich vor allem an Nasenspitze, Augenlidern, Glans penis oder Ohr, oft auch am Rumpf entlang der Hautspaltlinien. Charakteristisch sind kleine, himbeerrote Maculae, die in braune Plaques und rote Tumoren übergehen. Ein Befall der Schleimhäute und innerer Organe kommt vor.
Iatrogenes KS
Beim iatrogenen KS kommt ein Befall von Haut, Schleimhaut und inneren Organen vor. Der Verlauf ist aggressiver als bei der klassischen Form. Andererseits bilden sich die Erscheinungen nach Absetzen oder Wechsel der immunsupprimierenden Medikation meist zurück.
KS bei MSM
Beim KS bei MSM ohne HIV-Infektionen wird ein meist indolenter Verlauf mit kutanem Befall beschrieben. Prädilektionsstelle ist die Genitalregion.
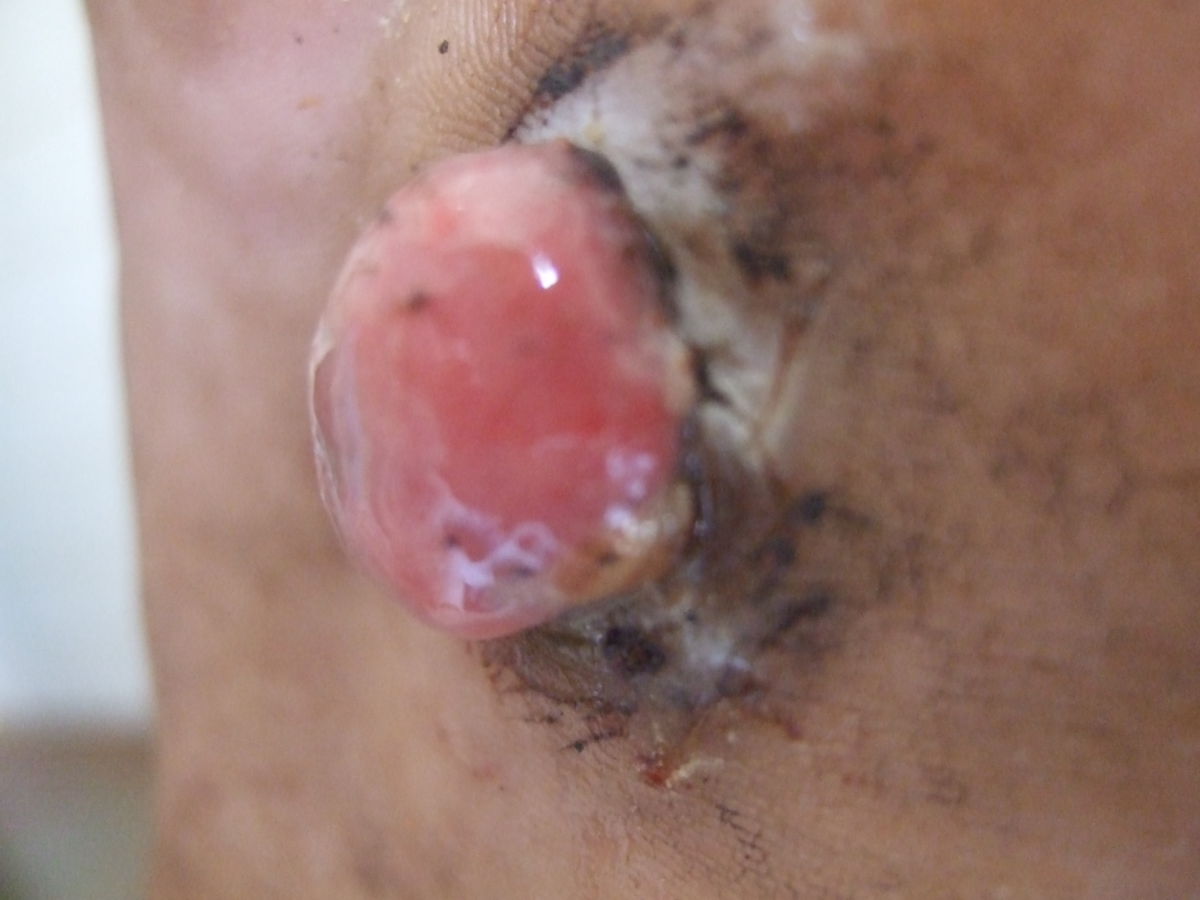
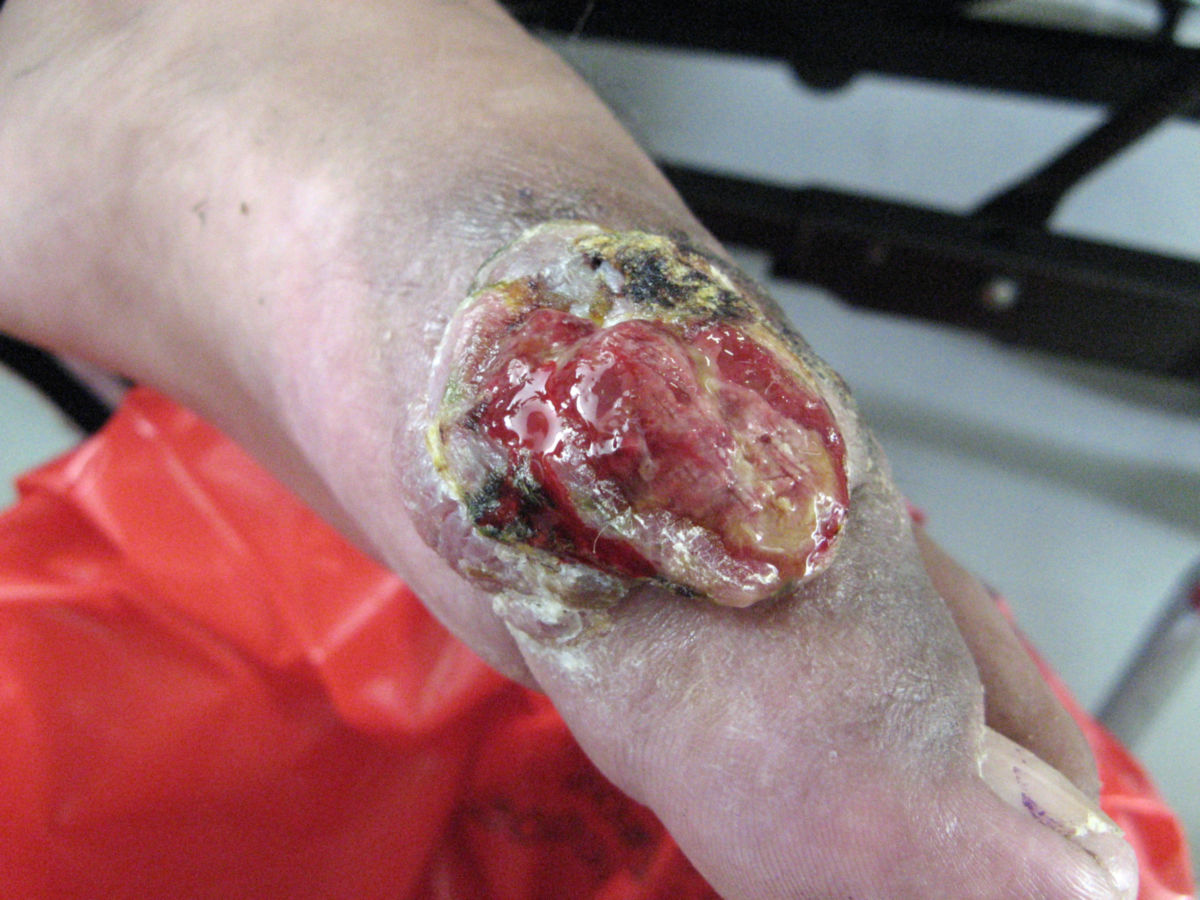
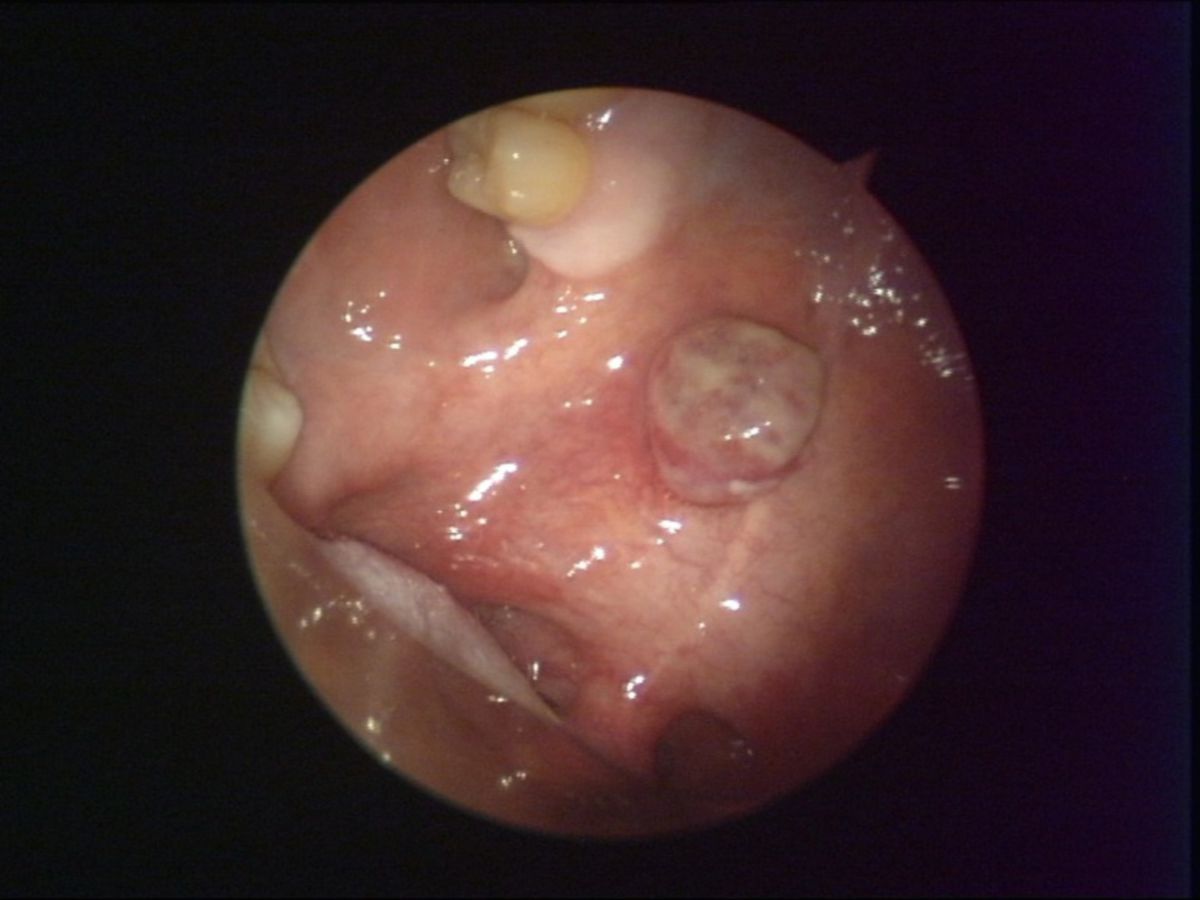
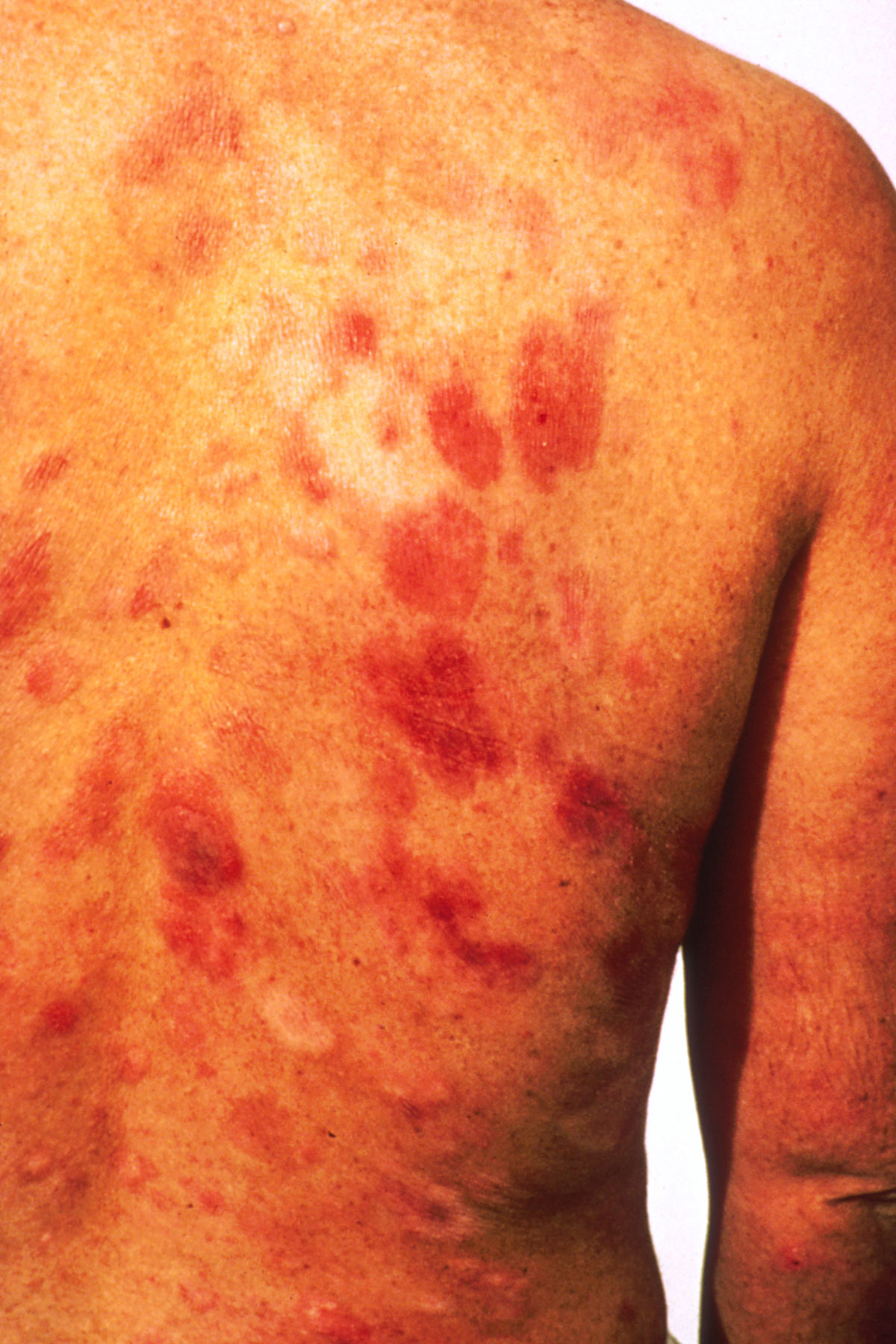
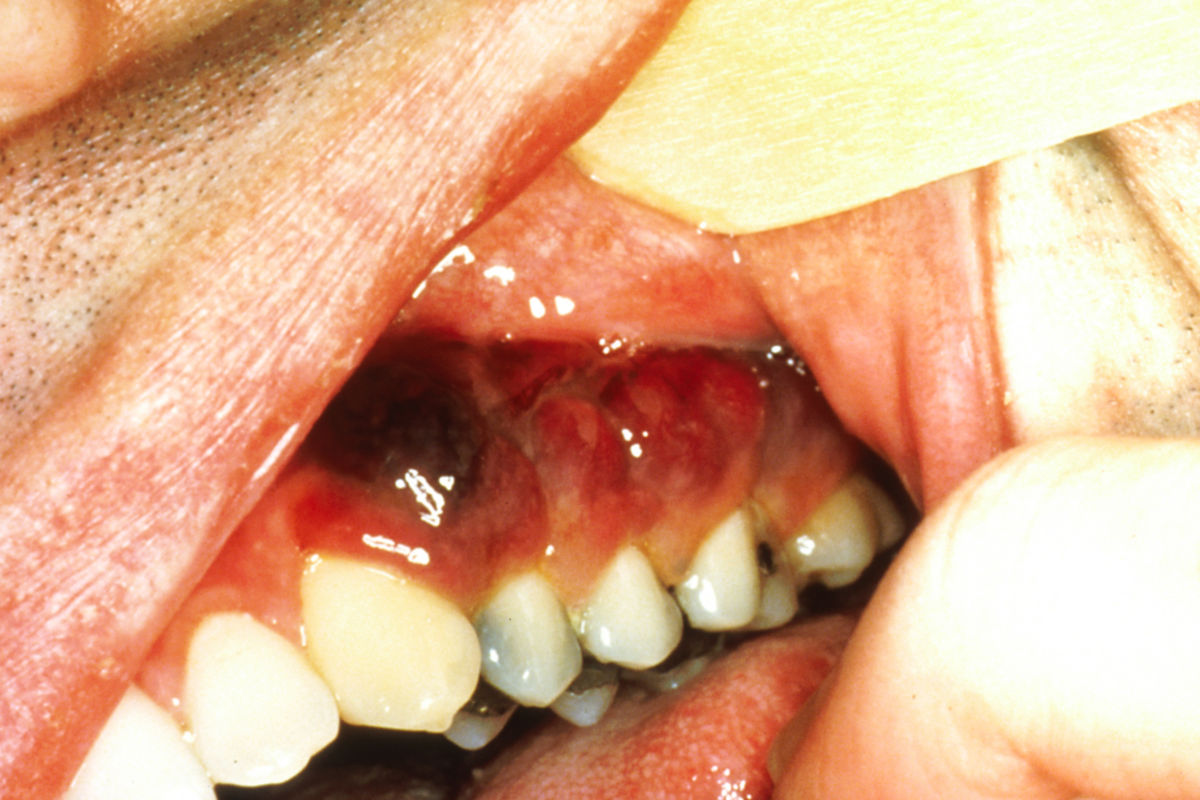
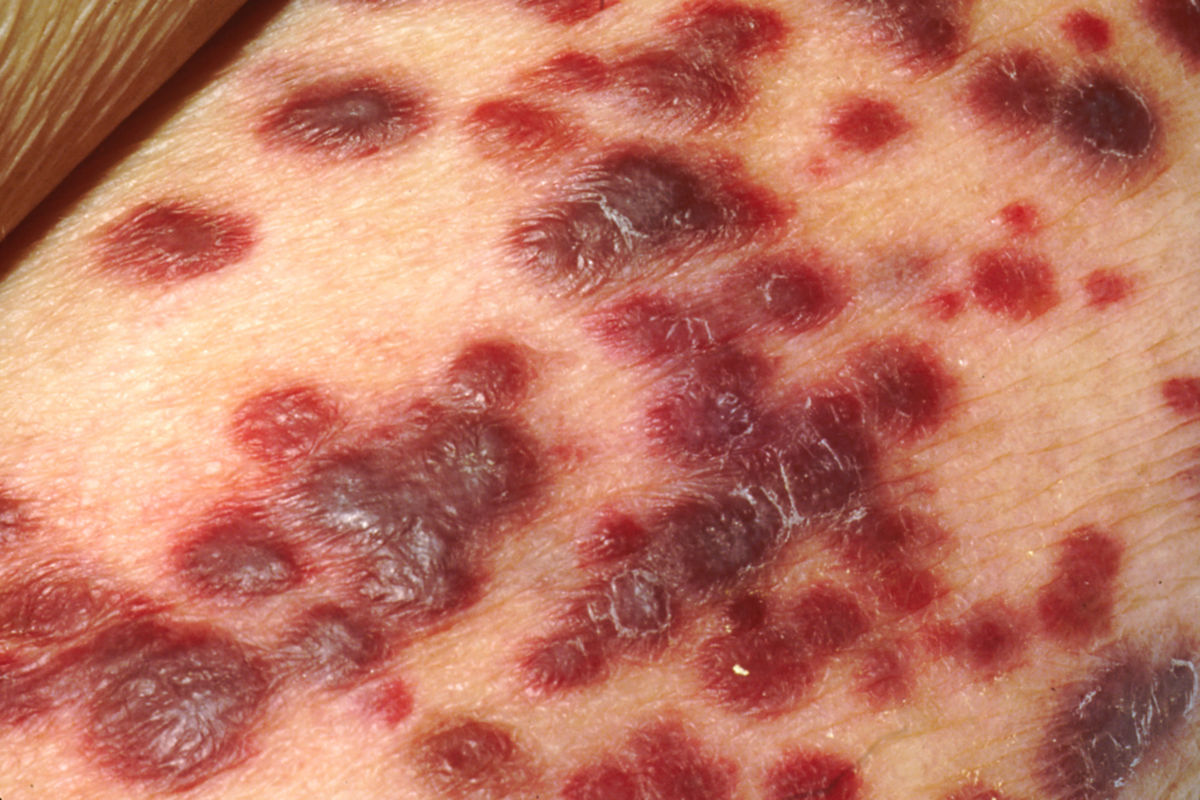
Stadien
Es existiert keine allgemein akzeptierte Stadieneinteilung.
...nach Mitsuyasu und Groopman
Die Stadieneinteilung nach Mitsuyasu und Groopman berücksichtigt die Ausbreitung und die Allgemeinsymptome:
- Stadium I: umschrieben kutan (< 10 Herde oder eine anatomische Region)
- Stadium II: disseminiert kutan (> 10 Herde oder > 1 anatomische Region)
- Stadium III: ausschließlich viszeral
- Stadium IV: kutan und viszeral
- Stadium IVA: ohne Allgemeinsymptomatik
- Stadium IVB: mit Fieber und/oder Gewichtsverlust
...nach ACTG
Die AIDS Clinical Trials Group (ACTG) entwickelte 1988 das TIS-Klassifikationssystem für HIV-assoziierte Kaposi-Sarkome:
- Tumor (T):
- T0: lokalisiert (z.B. nur in der Haut und/oder Lymhpknoten)
- T1: ausgedehnt (Ödem, ausgedehnte orale Beteiligung und/oder viszerale Beteiligung außer Lymphknoten)
- Immunsystem (I):
- I0: CD4-Zellzahl ≥ 200 Zellen/mm³
- I1: CD4-Zellzahl < 200 Zellen/mm³
- Systemischer Status (S):
- S0: keine systemische Erkrankung (keine opportunistischen Infektionen oder Soor in der Annamnese, keine B-Symptomatik, Karnofsky-Index ≥ 70)
- S1: systemische Erkrankung (Vorgeschichte von opportunistischen Infektionen oder Soor, mindestens ein B-Symptom, Karnofsky-Index < 70 und/oder Vorhandensein einer anderen HIV-bedingten Erkrankung, z.B. einer neurologischen Erkrankung oder eines Lymphoms)
Diagnostik
Die Verdachtsdiagnose eines Kaposi-Sarkoms wird klinisch gestellt. Die Diagnosebestätigung erfolgt primär durch histopathologische Untersuchung nach tiefer Biopsie. Allen Patienten sollten bei Erstdiagnose ein HIV-Test angeboten werden. Bei vermutetem oder bekanntem viszeralen Befall wird eine Ganzkörper-Computertomographie (CT) durchgeführt. Je nach Befund kommen z.B. eine Ösophagogastroduodenoskopie, eine Koloskopie und eine Bronchoskopie in Frage.
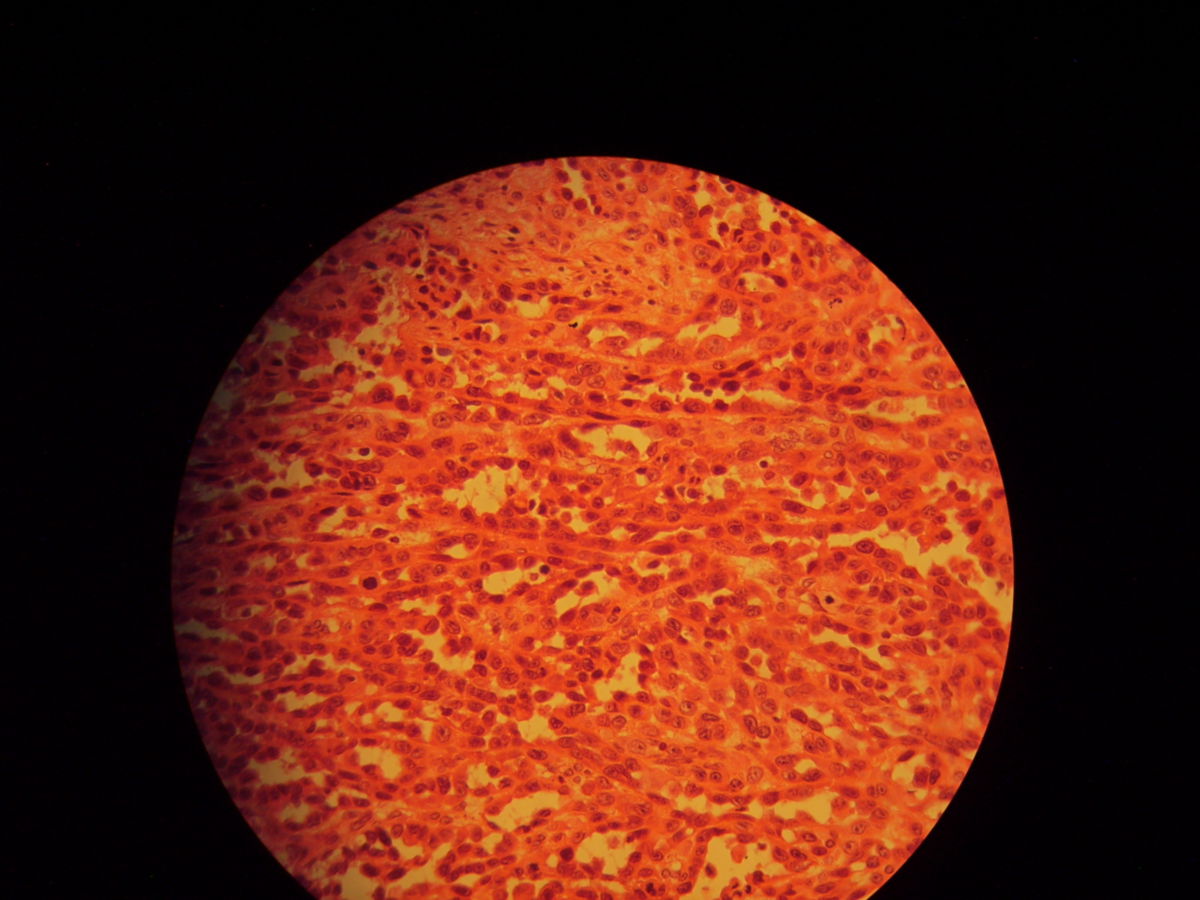
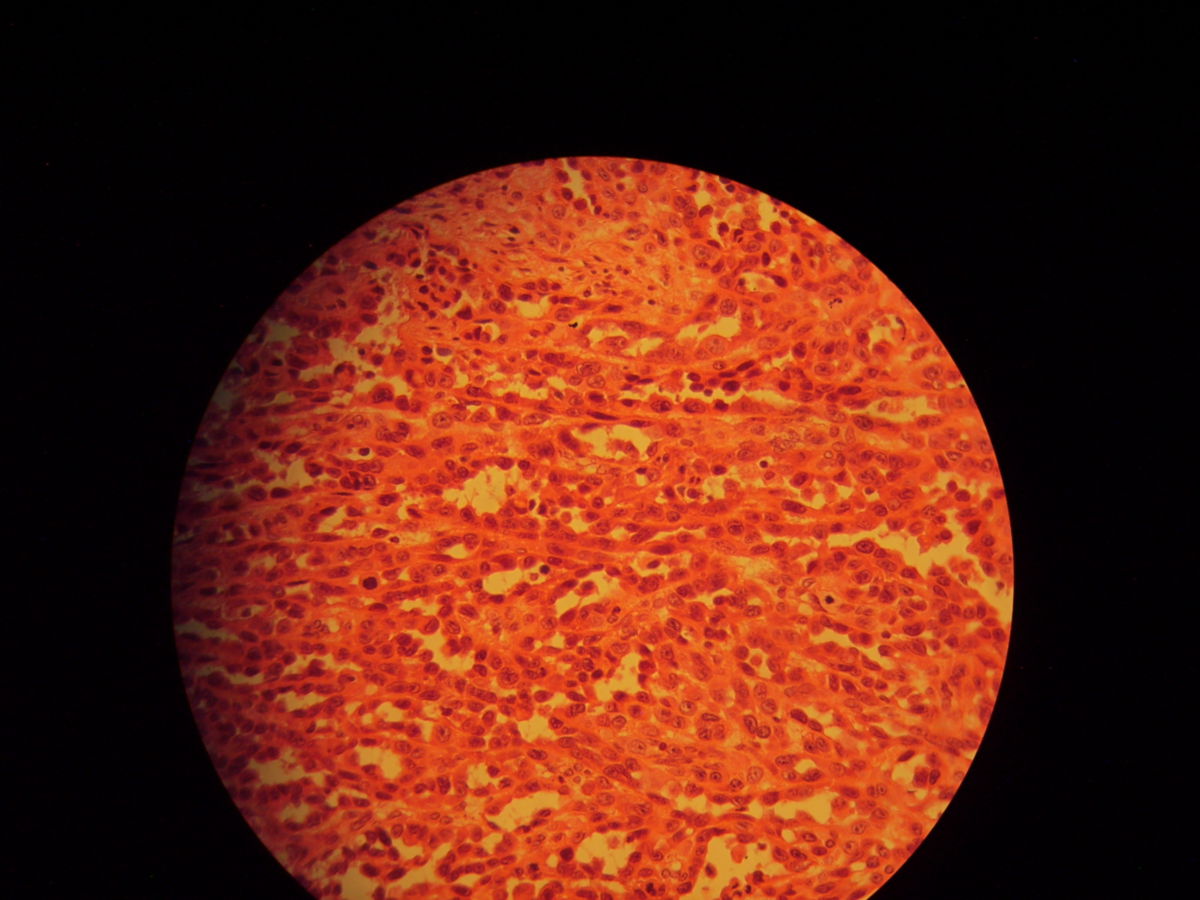
Histopathologie
- Fleckstadium: infiltrierende, Endothel-ausgekleidete, blutleere Spalten, oft um Adnexe und Gefäße (Promontoriumzeichen). Keine Zellatypien. Hämosiderin, Siderophagen und Plasmazellen sind typisch (pseudogranulomatöses Muster).
- Plaquestadium: gemeinsames Auftreten von kleinen Spindelzellfaszikeln, blutgefüllten Schlitzen und dilatierten Gefäßen
- Noduläres bzw. Tumorstadium: mitosereiche Spindelzellknoten ohne signifikante Kernatypien. Intrazytoplasmatische hyaline Globi.
Die Tumorzellen exprimieren CD31, CD34 und den lymphatischen Endothelmarker Podoplanin. Molekularbiologisch können HHV-8-DNA oder immunhistochemisch HHV-8-positive Zellkerne nachgewiesen werden.
Radiologie
Der radiologische Befund bei einem Kaposi-Sarkom ist meist unspezifisch und abhängig von der Tumorlokalisation.
siehe Hauptartikel: Kaposi-Sarkom (Radiologie)
Differenzialdiagnosen
Klinisch
Klinische Differenzialdiagnosen sind unter anderem:
- Angiome
- Granuloma teleangiectaticum
- Hämatome
- Angiokeratome
- Angiosarkome
- Dermatofibrome
- dermale melanozytäre Nävi
- Lymphome
- Lues II
- kutane T- oder B-Zell-Lymphome
- bazilläre Angiomatose
Histopathologisch
Histopathologische Differenzialdiagnosen sind z.B.
- progressives Lymphangiom (benignes Lymphangioendotheliom)
- targetoides hämosiderotisches Hämangiom
- mikrovenuläres Hämangiom
- aneurysmatisches fibröses Histiozytom
- Akroangiodermatitis Mali (Pseudo-Kaposi)
- Spindelzellhämangiom
Therapie
Je nach Klinik werden lokale oder systemische Therapien eingesetzt. Bei ausschließlich kutanem Befall mit einzelnen stabilen, asymptomatischen Läsionen kann auf eine Therapie verzichtet werden. Ausgedehnte Schleimhautläsionen, Lymphknotenbeteiligung, viszeraler Befall sowie rasch progrediente bzw. disseminierte Verläufe machen eine systemische Behandlung notwendig. Bei HIV-Infektion sollte umgehend eine ART eingeleitet oder optimiert werden. Bei iatrogenem KS wird die Immunsuppression modifiziert oder abgesetzt.
Lokaltherapeutische Optionen sind beispielsweise:
- Kryotherapie
- Alitretinoin
- Vincristin intraläsional
- Elektrochemotherapie
- Strahlentherapie
- Exzision einzelner Herde (in Einzelfällen)
Systemische Therapiemöglichkeiten sind:
Da eine Heilung in der Regel nicht möglich ist, ist das Ziel der Behandlung die Kontrolle über den Krankheitsverlauf und die Reduktion der Symptome. Man unterscheidet:
- Komplettes Ansprechen (Complete Response):
- Komplettes Verschwinden der Läsionen für mindestens 4 Wochen
- Partial Response:
- 50 % oder mehr Rückgang der Zahl der Läsionen
- 50% oder mehr Rückgang der Größe der Läsionen bzw. Rückgang der Schichtdicke
- Keine neuen Läsionen, mehr als 25 % Größenzunahme oder Ödemzunahme
- Stable disease:
- Jedes Ansprechen, das nicht die oben genannten Dimensionen erreicht
- Progressive Disease:
- Neue Läsionen
- Größenzunahme, Volumen-/Dickenzunahme vormals flacher Läsionen
- Zunahme des assoziierten Ödems
Leitlinie
- AWMF S1-Leitlinie, Stand 2021, abgerufen am 13.10.2022
Literatur
- Jary A et al. Kaposi's Sarcoma-Associated Herpesvirus, the Etiological Agent of All Epidemiological Forms of Kaposi's Sarcoma, Cancers (Basel). 2021 Dec 9;13(24):6208
- Restrepo CS et al. Imaging manifestations of Kaposi sarcoma, Radiographics. 2006 Jul-Aug;26(4):1169-85
- Vincenzi B et al. Classic Kaposi Sarcoma: to treat or not to treat?, BMC Res Notes. 2015 Apr 10;8:138
- Schneider JW, Dittmer DP Diagnosis and Treatment of Kaposi Sarcoma, Am J Clin Dermatol. 2017 Aug;18(4):529-539
- Cesarman E et al. Kaposi sarcoma, Nat Rev Dis Primers. 2019 Jan 31;5(1):9
Quellen
- Kaposi-Sarkom in DermIS (mit Abbildungen)
- Karger.com - Ein Fall von Sarcoma idiopathicum multiplex haemorrhagicum (Morbus Kaposi), zuletzt abgerufen am 16.01.2024