Brugada-Syndrom









Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
Loslegennach den Erstbeschreibern Pedro und Josep Brugada (1992)
Synonym: Brugada-Brugada-Syndrom
Abk: BRS, BrS
Englisch: Brugada syndrome
Definition
Das Brugada-Syndrom, kurz BrS, gehört zur Gruppe der kongenitalen Ionenkanalerkrankungen (Kanalopathie) des Herzens.
Epidemiologie
Ätiopathogenese
Die Ursache des Brugada-Syndroms ist erblich bedingt und noch nicht vollständig geklärt. In etwa 20 bis 25 % der Erkrankungsfälle liegt eine autosomal-dominante Punktmutation des SCN5A-Gens zu Grunde, das auf dem kurzen Arm von Chromosom 3 (3p21) lokalisiert ist. Es kodiert für die spannungsabhängigen kardialen Natriumkanäle (NaV). Die Loss-of-Function-Mutation verursacht einen strukturellen Defekt verschiedener Kanalkomponenten, die einen pathologisch verminderten Natriumstrom zur Folge haben. Hieraus resultiert eine zu frühe inhomogene Repolarisation der Kardiomyozyten, die durch Nachdepolarisationen ektope ventrikuläre Tachykardien mit z.T. letaler Folge verursachen kann.
Pathophysiologisch ist das Brugada-Syndrom vom Typ 3 des Long-QT-Syndroms (LQT3) abzugrenzen, bei dem die SCN5A-Mutation zu einer verzögerten Inaktivierung der NaV-Kanäle mit verlängerter Plateauphase im kardiomyozytären Aktionspotential führt. Entsprechend ist die Repolarisation verlängert.
Klinik
Das Brugada-Syndrom ist durch paroxysmale ventrikuläre Tachykardien (insbesondere Torsades de pointes) charakterisiert, die mit folgenden Symptomen einhergehen können:
- Synkopen
- pektanginösen Beschwerden
- allgemeinem Unwohlsein und
- Schweißausbrüchen
Komplikationsbedingt können die Herzrhythmusstörungen zu Kammerflimmern bis hin zum Herzstillstand degenerieren.
Die klinische Symptomatik tritt meist erst im 3. bis 5. Lebensjahrzehnt auf, Kinder können unauffällig sein.
Diagnostik
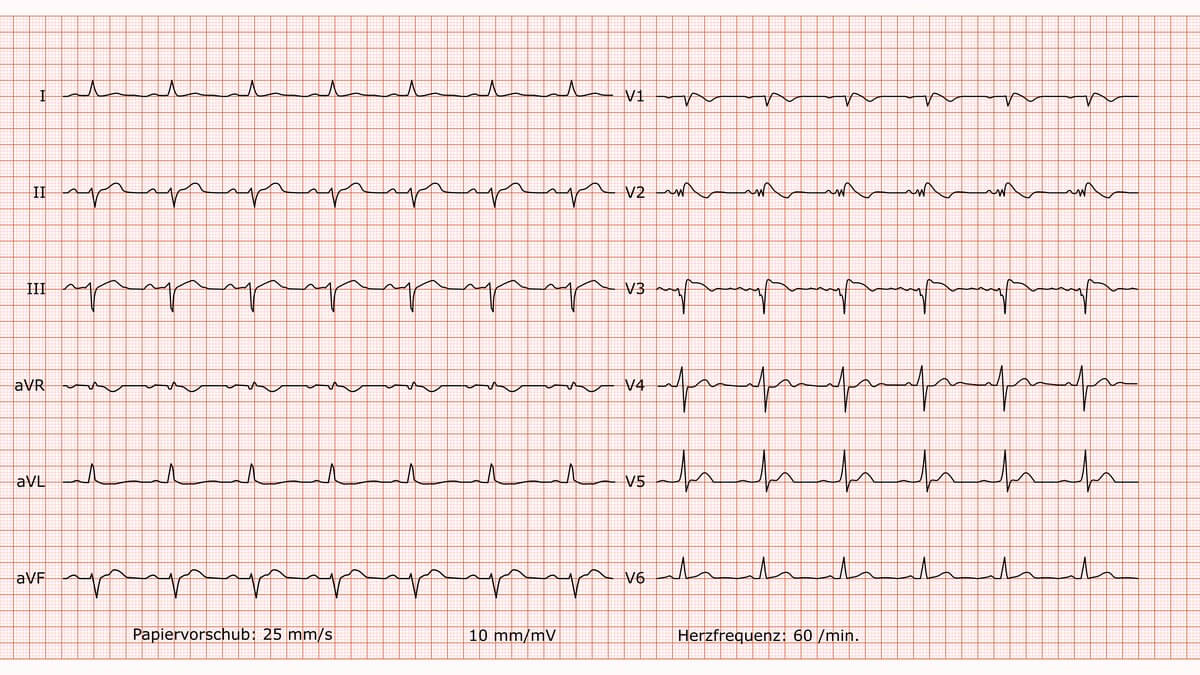
EKG
Die Diagnose des Brugada-Syndroms ist komplex. Die EKG-Veränderungen sind intraindividuell sehr variabel und bei einigen Patienten gar nicht bzw. nur zeitweise zu beobachten. Im Ruhe-EKG zeigen sich:
- kompletter oder inkompletter Rechtsschenkelblock und
- gewölbte ST-Strecken-Hebungen bzw. J-Wellen in den rechts präkordialen Ableitungen (V1-V3) mit anschließender T-Negativierung. Ohne diese T-Negativierung liegt ein Pseudo-Brugada-Muster vor.
Neben diesem (diagnostischen) Typ 1 gibt es noch Typ 2 und 3, die jeweils mit sattelförmigen ST-T-Komplexen einhergehen. Da diese EKG-Merkmale auch bei trainierten Ausdauersportlern mit linksventrikulärer Hypertrophie vorkommen können, gelten Typ 2 und 3 als nicht diagnostisch.
Im Rahmen des stummen bzw. maskierten Krankheitstypus zeigt sich ein normgerechtes EKG-Bild, das unter oraler Gabe von Klasse-I-Antiarrhythmika (Natriumkanalblocker) entsprechend umschlägt, d.h. demaskiert wird. Aufgrund der hohen Komplikationsdichte der Provokationstests (z.B. Ajmalin-Test) bedarf es eines begleitenden EKG-Monitorings mit Defibrillator-Bereitschaft. Bei Patienten ohne Veränderungen der ST-Strecke nach Gabe eines Natriumkanalblockers ist ein Brugada-Syndrom sehr unwahrscheinlich.
Als weiteres diagnostisches Verfahren besteht die Möglichkeit der elektrophysiologischen Untersuchung (EPU) zur Ableitung des intrakardialen EKGs.
Molekulardiagnostik
Bei bekannten Krankheitsfällen in der Familienanamnese ist ein molekulargenetischer Nachweis der auslösenden SCN5A-Mutation sinnvoll. Er erfolgt in einem entsprechend ausgerüsteten Labor mittels Sanger-Sequenzierung oder NGS. Der Nachweis gelingt aber nur in etwa 20 bis 25 % der Fälle.
Therapie
Da es keine adäquate medikamentöse Therapie des Brugada-Syndroms gibt, besteht die Standardtherapie in der Implantation eines AICD.
Cave: Der Einsatz von Beta-Blockern bei vorliegendem Brugada-Syndrom ist kontraindiziert. Eine ventrikuläre Tachykardie im Rahmen eines Brugada-Syndroms tritt typischerweise aus einer Bradykardie heraus auf. Beta-Blocker senken die Herzfrequenz und können eine Bradykardie auslösen. Dadurch können sie das Risiko einer paroxysmalen ventrikulären Tachykardie erhöhen.[2][3]
Quellen
- ↑ Orphanet – Brugada Syndrom, aufgerufen am 24.5.2023
- ↑ Moog et al. Medizinische Genetik für die Praxis, Thieme Verlag, 2014
- ↑ Brodie et al. Pharmacological Therapy in Brugada Syndrome, Arrhythm Electrophysiol Rev, 2018