Vollständige Exomsequenzierung



Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenSynonyme: Gesamtexomsequenzierung, Exomsequenzierung
Englisch: whole exome sequencing, exome sequencing
Definition
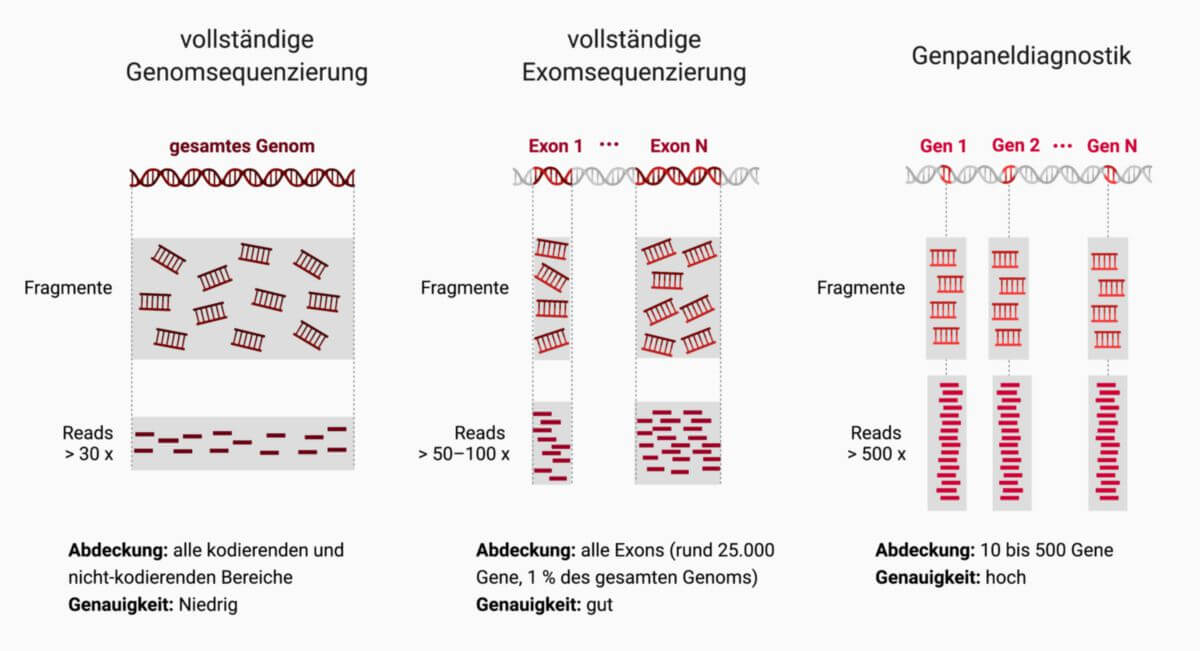
Die vollständige Exomsequenzierung, kurz WES, ist ein molekulargenetisches Analyseverfahren zur vollständigen Sequenzierung aller Exons eines Organismus. Das sogenannte Exom ist der Teil der DNA, der Proteine kodiert. Die WES dient als diagnostischer Ansatz zur Identifizierung genetisch bedingter molekularer Defekte bei Patienten.
siehe auch: Exom-Trio-Analyse, Genpaneldiagnostik
Hintergrund
Die Ursache für sporadisch auftretende Erkrankungen, die oft als Einzelfall in einer Familie vorkommen, ist meist eine Neumutation innerhalb eins Gens. In der OMIM-Datenbank (Online Mendelian Inheritance in Man) sind mehr als 4.600 Gene aufgelistet, deren Mutation einen – oft krankhaften – Phänotyp hervorruft.[1] Hierzu zählen viele seltene Entwicklungsstörungen sowie Fälle von Intelligenzminderung.
Der Vorteil im Vergleich zur vollständigen Genomsequenzierung (WGS) ist die Beschränkung auf die Protein-kodierenden Bereiche, die nur etwa 1 % des gesamten Genoms ausmachen. Dies führt zu einer nennenswerten Reduktion der Kosten und des Sequenzierungsaufwandes. Der Nachteil ist jedoch, dass beim WES durch die vorangehende Selektion und Anreicherung der Exons Artefakte entstehen können.
Ablauf
Der Ablauf des WES gliedert sich in folgende Schritte:
- Patientenvorbereitung (inkl. Probenentnahme)
- Exom-Anreicherung
- Sequenzierung
- bioinformatische Analyse
Patientenvorbereitung
Vor dem WES findet zunächst eine humangenetische Beratung des Patienten statt. Hierbei wird u.a. festgelegt, wie mit nicht indikationsbezogenen Informationen umgegangen wird, die aus dem WES hervorgehen. Eine Vorauswahl indikationsrelevanter Gene erfolgt basierend auf dem klinischen Erscheinungsbild. Die Symptome sollten dabei als HPO-Terms (Human Phenotype Ontology) vorliegen.
Nach der Beratung und Aufklärung erfolgt die Probenentnahme, meist werden Blutproben verwendet. Es sind jedoch auch Speichelproben möglich.
Exom-Anreicherung
Zur Anreicherung der Exons existieren zwei Methoden:
- Lösungsbasiertes Einfangen ("solution-based capture")
- Array-basiertes Einfangen ("array-based capture")
Zunächst wird die extrahierte DNA durch Ultraschall in Fragmente zerlegt, die klein genug sind, um sequenziert zu werden. Danach werden diese Fragmente mit einzelsträngigen Oligonukleotiden hybridisiert. Diese sind so aufgebaut, dass sie spezifisch nur die Exon-Sequenzen binden. Bei der Array-basierten Methode sind die Oligonukleotide auf einer Platte immobilisiert, bei der lösungsbasierten Methode an kleinen Kugeln ("beads"). Die ungebundenen DNA-Fragmente, welche die nicht-kodierenden Bereiche darstellen, werden ausgewaschen.
Sequenzierung
Zur Sequenzierung eignen sich verschiedene Next Generation Sequencing-Methoden, z.B. die Illumina- oder SOLiD-Sequenzierung. Bei der Sequenzierung wird die Basenabfolge (A, T, C, G) für jedes einzelne Fragment definiert.
Bioinformatik
Die Aufarbeitung der Sequenzierungsergebnisse ist komplex. Meist existieren etablierte Pipelines, welche die Fragmentsequenzen den entsprechenden Genen zuordnen ("mapping and alignment") und mutierte Allele bzw. Genvarianten identifizieren und herausfiltern. Bei einer Exom-Trio-Analyse werden zudem die Ergebnisse der Familienmitglieder miteinander verglichen.
Literatur
- Yang et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders N Engl J Med 2013
- MVZ Martinsried - Whole Exome Sequencing (WES) / Whole Genome Sequencing (WGS), abgerufen am 29.03.2022
- Universität Mainz - Vorlesung Genomforschung und Sequenzanalyse, abgerufen am 29.03.2022
Quellen
- ↑ OMIM - GeneMap, abgerufen am 29.03.2022