Pyruvatdehydrogenase







Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenSynonyme: Pyruvat-Dehydrogenase, PDH, Pyruvatdehydrogenase-Komplex
Englisch: pyruvate dehydrogenase
Definition
Die Pyruvatdehydrogenase, kurz PDH, ist ein Enzym, das die Reaktion von Pyruvat zu Acetyl-CoA katalysiert, und damit die Glykolyse an den Citrat-Zyklus anschließt.
Grundlagen
Die Pyruvatdehydrogenase setzt Pyruvat unter der Freisetzung von CO2 zu Acetyl-CoA um. Während dessen wird ein NAD+ zu NADH reduziert.
Der Reaktionsverlauf der Pyruvatdehydrogenase ist irreversibel – die Reaktion verläuft entsprechend nur in eine Richtung. Somit kann Acetyl-CoA nicht in Pyruvat bzw. Glucose umgesetzt werden. Es ist zwar möglich Kohlenhydrate zu Acetyl-CoA abzubauen und in weiterem Verlauf Fettsäuren zu synthetisieren, jedoch ist es nicht möglich, aus Fettsäuren Kohlenhydrate zu bilden.
Aufbau
Die Pyruvatdehydrogenase ist ein sogenannter Multienzymkomplex, der aus drei unterschiedlichen Enzymen besteht (E1-E3) und im Mitochondrium lokalisiert ist. Damit eine Katalyse ihrer Reaktionen vollständig durchgeführt werden kann, benötigt die PDH insgesamt fünf Coenzyme.
| Enzymkomponente | Coenzyme | Beschaffenheit des Coenzyms |
|---|---|---|
| Pyruvat-Dehydrogenase (E1) | Thiaminpyrophosphat (TPP) = aktiviertes Thiamin | enzymgebunden (keine kovalente Bindung) |
| Dihydroliponamid-Acetyltransferase (E2) | Liponsäure (Liponamid) | enzymgebunden (kovalent: Amidbindung an einem Lysinrest von E2, deshalb "Liponamid" |
| Coenzym A | löslich | |
| Dihydroliponamid-Dehydrogenase (E3) | FAD | enzymgebunden (fest, aber nicht kovalent) |
| NAD+ | löslich |
Die oben aufgeführten drei Enzymkomponenten (E1, E2, E3) sind:
- E1: Pyruvatdehydrogenase: Dieses Enzym bindet das Pyruvat und katalysiert mit der Hilfe des Coenzyms Thiaminpyrophosphat (TPP) die Decarboxylierung von Pyruvat. Es entsteht dabei CO2.
- E2: Dihydroliponamid-Acetyltransferase: E2 katalysiert vor allem den Transfer des Acetylrests, der vom Pyruvat übrig geblieben ist, auf das Coenzym A (CoA). Während dieses Vorgangs werden zwei Schwefelatome ihres Coenzyms Liponsäure (Liponamid) zu SH-Gruppen reduziert.
- E3: Dihydroliponamid-Dehydrogenase: Dieses Enzym nimmt mit Hilfe des Coenzyms (FAD) die Elektronen der beiden SH-Gruppen des Liponamids auf und überträgt sie anschließend auf NAD+. Dieser Vorgang ermöglicht die Regeneration von Liponamid.
Neben diesen drei Enzym-Untereinheiten enthält die PDH noch zwei regulatorische Untereinheiten – diese können die PDH je nach Bedarf an- bzw. abschalten.
Reaktionsschritte
Überblick
- Pyruvat (Endprodukt der Glykolyse) wird unter Abspaltung von CO2 auf das Thiaminpyrophosphat (Coenzym von E1) übertragen, wobei ein Hydroxyethylrest entsteht (= aktivierter Acetaldehyd).
- Der entstandene Acetaldehyd wird von Thiaminpyrophosphat (E1) auf ein Liponamid (Coenzym von E2) übertragen und gleichzeitig zu einer Acetylgruppe oxidiert.
- Das Liponamid überträgt anschließend die Acetylgruppe auf das Coenzym A, so dass Acetyl-CoA entsteht. Wichtig ist, dass dabei die Disulfidgruppe des Liponamids in zwei einzelne SH-Gruppen umgewandelt wird.
- Da das Elektron jeder SH-Gruppe unter Vermittlung von E3-gebundenem FAD an NAD+ abgegeben wird, regeneriert die Disulfidgruppe des Liponamids. Zusätzlich wird dabei NADH gebildet.
Pyruvatdehydrogenase ist sowohl für die Decarboxylierung des Pyruvats, als auch für die Oxidation des aktivierten Acetaldehyds als Katalysator zuständig. Deshalb wird die Gesamtreaktion als oxidative Decarboxylierung von Pyruvat bezeichnet.
Schritt 1
Das Thiaminpyrophosphat (TPP), das als Coenzym für die Pyruvat-Dehydrogenase (E1) fungiert, weist chemisch gesehen zwei heterozyklische Ringe auf. Für die Coenzym-Funktion ist der Thiazolring entscheidend. Der Kohlenstoff, der im Thiazolring zwischen dem Stickstoff- und Schwefelatom liegt, tendiert dazu, leicht ein Proton abzugeben. Es entsteht somit ein negativ geladenes und sehr reaktives Carbanion. Dieses Carbanion leitet die PDH-Reaktion ein, indem es sich an den Carbonylkohlenstoff des Pyruvat anlagert und folglich auf die Elektronen am Pyruvat einen kräftigen Elektronenzug ausübt. Im weiteren löst sich die Carboxylgruppe in Form von CO2 ab. Zurück bleiben zwei Elektronen der Carboxylgruppe am TPP.
Parallel zu dieser Reaktion lagert sich ein Proton an den Sauerstoff der Carbonylgruppe (–C=O) an. Dadurch bildet sich eine Doppelbindung zu TPP aus und es entsteht Hydroxyethyl-TPP. Dieser Hydroxyethylrest wird auch als "aktivierter Acetaldehyd" bezeichnet, da die Oxidationsstufe des Carbonylkohlenstoffs in Hydroxyethyl-TPP der Oxidationsstufe des C-Atoms im Acetaldehyd entspricht.
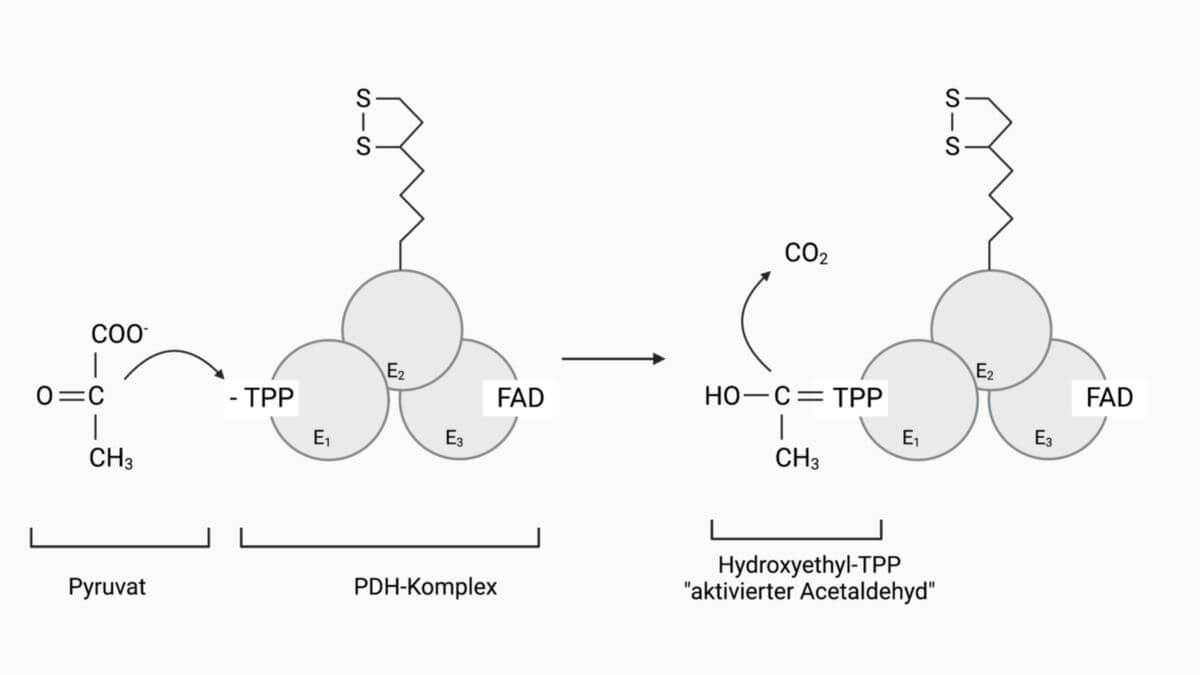
Schritt 2
Der aktivierte Acetaldehyd wird im 2. Schritt von Thiaminpyrophosphat auf das Liponamid – eine fest gebundene prosthetische Gruppe der Dihydroliponamid-Acetyltransferase (E2) – übertragen:
- Es öffnet sich die Disulfidgrupe des Liponamids, die aufgrund des Transfers im oxidierten Zustand vorliegt.
- Da zwei Schwefelatome vorhanden sind, lagert sich Acetaldehyd an eines der beiden. Das verbliebene Schwefelatom nimmt zusammen mit einem Proton die beiden überzähligen Elektronen auf, die während der Abspaltung (voriger Schritt) des CO2 am TPP zurück geblieben sind.
In diesem Moment wird Acetaldehyd (C2H4O) zu einer Acetylgruppe (–C(O)CH3) oxidiert. In der Acetylgruppe entspricht die Oxidationsstufe des Carbonylkohlenstoffs der Oxidationsstufe des äquivalenten C-Atoms in Acetat. In diesem Reaktionsschritt wird folglich Acetaldehyd zu Acetat oxidiert. Allerdings liegt das Acetat nicht frei sondern in Form eines Thioesters vor.
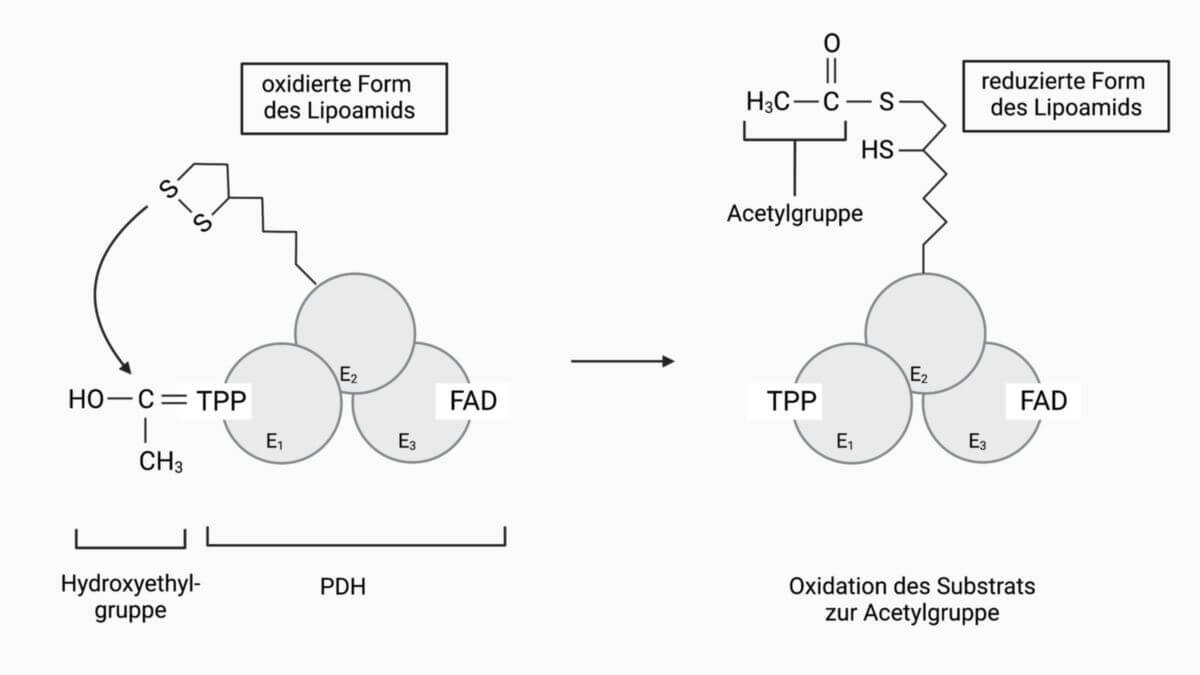
Schritt 3
Chemisch betrachtet ist das Liponamid ein lang gestrecktes Molekül – bildhaft als "langer Arm" vorstellbar. Es hat die Funktion, die Acetylgruppe auf das Coenzym A (CoA) zu übertragen. Hierbei entsteht Acetyl-CoA (aktivierte Essigsäure) an der Dihydroliponamid-Acetyltransferase (E2). Anstelle der ursprünglichen Disulfidgruppe enthält das Liponamid jetzt zwei SH-Gruppen.
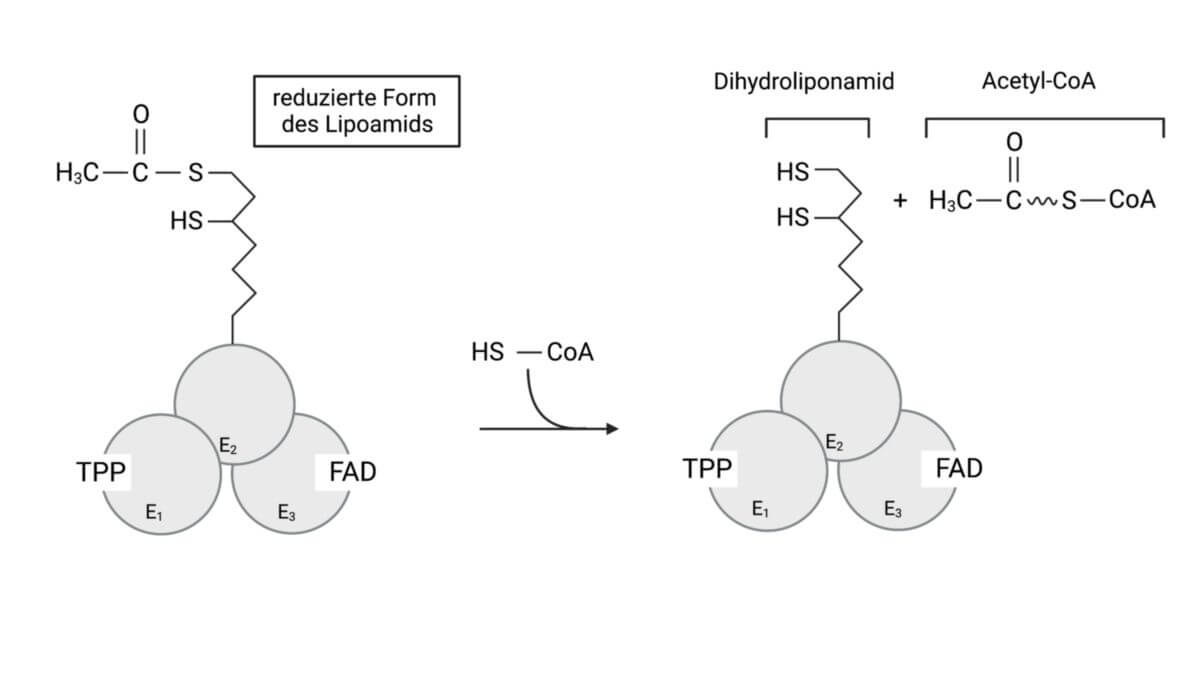
Schritt 4
Damit das Liponamid seine beiden SH-Gruppen oxidieren kann, um die Disulfidbindung zu regenerieren, schwenkt es seinen "Arm" zur Dihydroliponamid-Dehydrogenase (E3). Durch dieses Vorgehen werden beide SH-Gruppen durch die Reaktion der prosthetischen Gruppe FAD oxidiert. Als Resultat erhält man FADH2 – dieses gibt beide übertragenen Elektronen an NAD+ weiter. Folglich enthält das Reaktionsprodukt (NADH) beide überzähligen Elektronen – beide waren ursprünglich bei der Abspaltung des CO2 am TPP zurückgeblieben. Das Liponamid steht somit einem neuen Reaktionszyklus zur Verfügung.
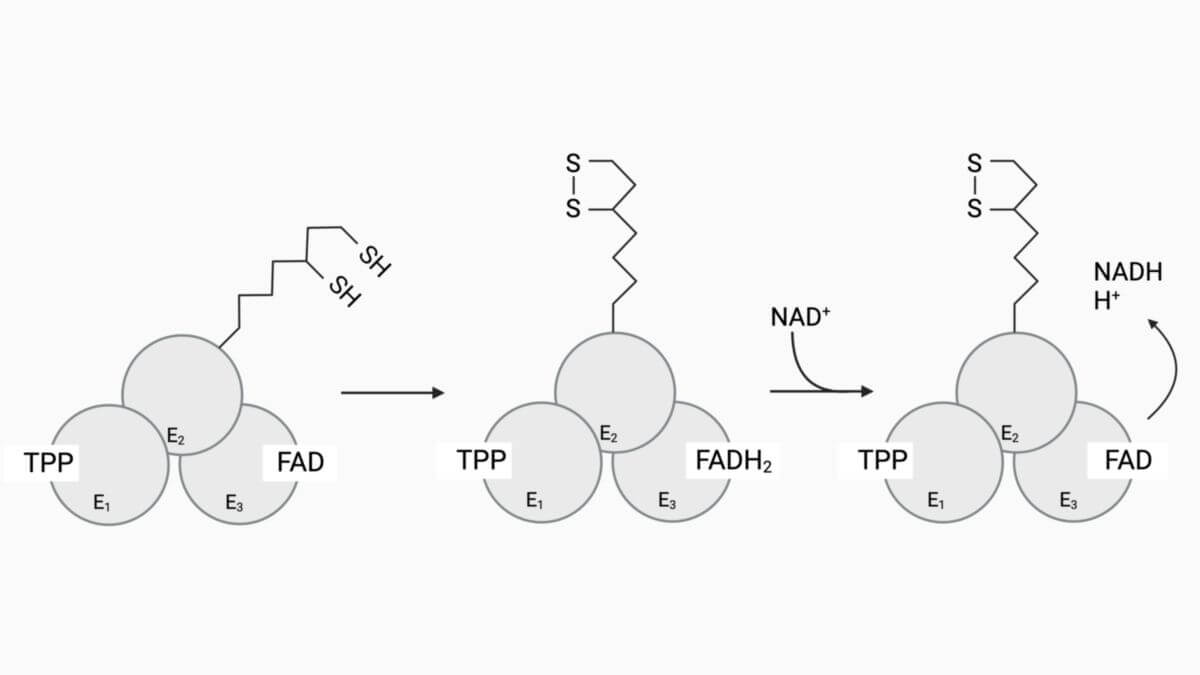
Bilanz
Während des ganzen Reaktionszyklus der Pyruvatdehydrogenase entsteht ein Molekül CO2, ein Acetyl-CoA und ein NADH.
Regulation
Die Pyruvatdehydrogenase befindet sich in der Matrix, d.h. im Innenraum der Mitochondrien und kann nicht in der gleichen Weise reguliert werden wie Enzyme des Zytosols. Die mitochondriale Membran ist nicht für cAMP permeabel. Außerdem sind Phosphorylierungen, die in den Mitochondrien ablaufen, im Vergleich zum Zytosol von geringer Bedeutung.
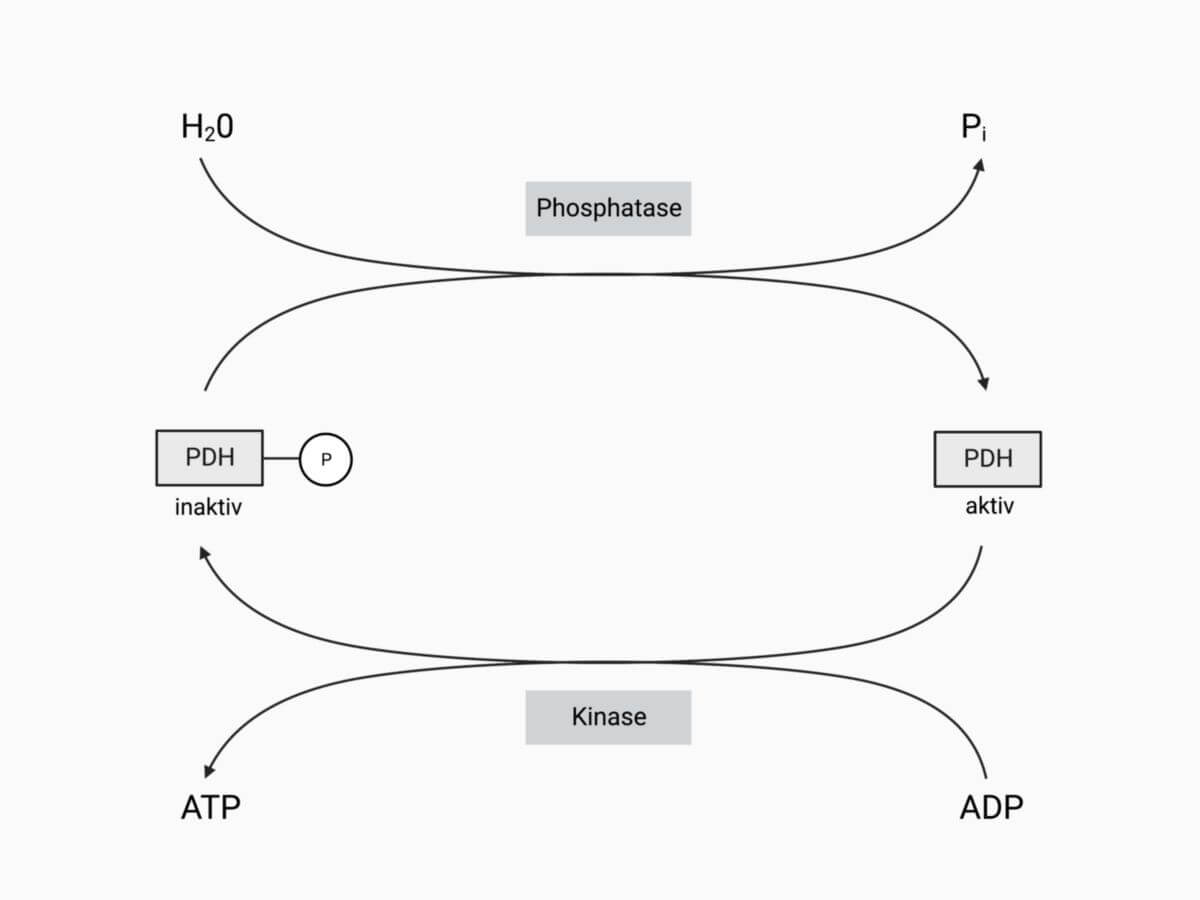
Die Aktivität der Pyruvatdehydrogenase wird durch eine reversible Phosphorylierung gesteuert: Sind hinreichende Mengen an Acetyl-CoA und NADH im Mitochondrium vorhanden, wird die Pyruvatdehydrogenase durch die Phosphorylierung abgeschaltet. Dies geschieht, da hohe Pyruvatkonzentrationen die Phosphorylierung unterbinden. Das PDH bleibt so im aktiven Zustand und das angesammelte Pyruvat kann verarbeitet werden.
Diese Hemmung kommt dadurch zustande, dass eine Kinase durch Acetyl-CoA und NADH stimuliert wird. Darauf folgt eine Phosphorylierung der E1-Untereinheit des Enzyms an einem bestimmten Serinrest. Folglich unterdrücken hohe Pyruvat-Konzentrationen die Aktivität der Kinase.
Soll das Enzym wieder aktiviert werden, wird – durch eine Phosphatase – die inaktivierende Phosphatgruppe am Serinrest der E1-Untereinheit abgespalten. Die Phosphatase ist ebenfalls ein Bestandteil des PDH-Multienzymkomplexes. Da die Phosphatase von Calcium-Ionen abhängig ist, vermutet man, dass die mitochondriale Calciumkonzentration einen Einfluss auf die Aktivität der PDH hat.
Acetyl-CoA und NADH vermitteln an der Pyruvatdehydrogenase eine klassische Produkthemmung. Sind sie in ausreichenden Mengen an den Mitochondrien akkumuliert, blockieren sie an den Untereinheiten der PDH die Bindestellen für Coenzym A und auch für NAD+.
Klinische Bedeutung
Ein Mangel an einer der Komponenten des PDH-Komplexes ist ursächlich für die PDH-Zytopathie.