Ceroidlipofuszinose











Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenSynonyme: NCL, CLN, neuronale Ceroidlipofuszinose
Englisch: ceroid lipofuscinosism, neuronal ceroid lipofuscinosis
Definition
Neuronale Ceroidlipofuszinosen, kurz NCLs, sind eine heterogene Gruppe progressiver, neurodegenerativer, erblicher Erkrankungen, die u.a. mit Demenz, Epilepsie und Sehverlust einhergehen. Sie werden den lysosomalen Speichererkrankungen zugeordnet. Es existieren verschiedene Formen der Erkrankung, die in den meisten Fällen autosomal-rezessiv vererbt werden.
Epidemiologie
Betroffen ist etwa eines von etwa 30.000 Neugeborenen. Damit sind die neuronalen Ceroidlipofuszinosen die häufigste Gruppe neurodegenerativer Krankheiten, die im Kindes- und Jugendalter ihren Anfang nehmen.
In Deutschland treten am häufigsten die infantile CLN1, die spätinfantile CLN2 und die juvenile CLN3 auf.
Genetik
Bisher sind 13 Typen der Erkrankung bekannt, die alle auf Mutationen in verschiedenen Genen beruhen. Die Vererbung verläuft autosomal-rezessiv. Eine Ausnahme stellt die adulte Form CLN4 dar, die autosomal-dominant vererbt wird.
Pathophysiologie
Durch die spezifischen Genmutationen kommt es zu Einschlüssen von fluoreszierenden Lipopigmenten, sogenanntem Ceroid, in den Lysosomen zahlreicher Gewebe. Das Ceroid ähnelt dem auch als Alterspigment bezeichneten Lipofuszin. Durch die Einschlüsse werden in erster Linie die Neuronen der grauen Substanz sowie der Netzhaut geschädigt. Daraus resultiert eine ausgedehnte neuronale Entzündungsreaktion und Degeneration im ZNS.
Symptome und Verlauf
Neuronale Ceroidlipofuszinosen haben gemeinsam, dass die Betroffenen initial vollkommen gesund erscheinen, mit Ausnahme der kongenitalen Form CLN10.
Das Alter bei Krankheitsbeginn kann je nach Form von der Geburt bis zum Erwachsenenalter reichen.
Die Hauptsymptome sind Visusverlust, Rückschritte in der geistigen und motorischen Entwicklung, Demenz sowie Epilepsie.
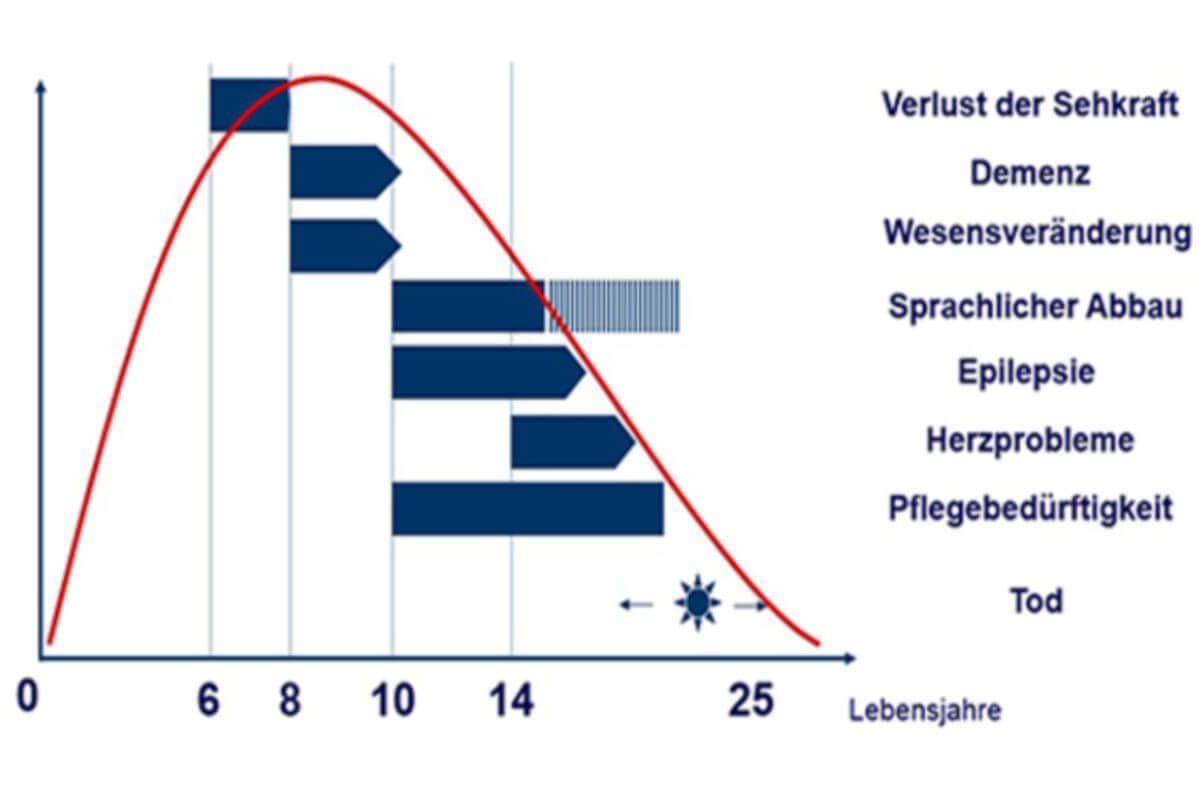
Klassifikation
Aktuell (2023) werden 13 verschiedene Typen der Erkrankung unterschieden:
| Typ | Bezeichnung | betroffenes Gen | Manifestationsalter | Merkmale |
|---|---|---|---|---|
| CLN1 | infantile NCL | CLN1/PPT1 | Beginn mit 6 bis 18 Monaten, auch Erwachsenenalter | Vermindertes Schädelwachstum, Krampfanfälle, motorische Entwicklungsrückschritte |
| CLN2 | (klassische) spätinfantile NCL | TPP1 | Kleinkindalter, 2 bis 4 Jahre | Krampfanfälle, Ataxie, Visusverlust, verzögerte Sprachentwicklung, motorische Entwicklungsrückschritte |
| CLN3 | (klassische) juvenile NCL | CLN3 | Schulalter bzw. 4 bis 7 Jahre | Visusverlust, Verhaltensauffälligkeiten, kognitiver Abbau |
| CLN4 | adulte NCL, autosomal-dominant | DNAJC5 | Erwachsenenalter | unspezifische mentale oder motorische Symptome, Verhaltensauffälligkeiten |
| CLN5 | spätinfantile NCL | CLN5 | Kleinkindalter, 2 bis 5 Jahre | Lernstörung, kognitive Einschränkungen |
| CLN6 | spätinfantile NCL | CLN6 | Kleinkindalter, 2 bis 5 Jahre | Krampfanfälle, Ataxie, verzögerte Sprachentwicklung |
| CLN7 | spätinfantile NCL | CLN7/MFSD8 | Kleinkindalter, 2 bis 5 Jahre | Krampfanfälle, Visusverlust, motorische und kognitive Rückentwicklung |
| CLN8 | spätinfantile NCL | CLN8 | Kleinkind - Schulalter | Krampfanfälle, Visusverlust, motorische und kognitive Rückentwicklung |
| CLN10 | Kongenitale NCL | 'CLN10/CathepsinD (CTSD) | ab der Geburt, teilweise auch erst später | Mikrozephalie, Dysmorphien, Krampfanfälle, Hyperkinesien |
| CLN11 | adulte NCL | Progranulin (GRN) | ab 20. Lebensjahr | unspezifische mentale oder motorische Symptome, Verhaltensauffälligkeiten |
| CLN12 | adulte NCL | ATPase, Transmebran-Ionen Transporter (ATP13A2) | 2. bis 3. Lebensdekade | unspezifische mentale oder motorische Symptome, Verhaltensauffälligkeiten, Rigidität, Hypokinesie |
| CLN13 | adulte NCL | CathepsinF (CTSF) | ab 20. Lebensjahr | unspezifische mentale oder motorische Symptome, Verhaltensauffälligkeiten |
| CLN14 | progressiver MyETyp | CLN14 /Kaliumkanalprotein (KCTD7) | 1. Lebensjahr | vermindertes Schädelwachstum, Krampfanfälle (Myoklonus) |
Diagnostik
Der erste Arztkontakt findet meist aufgrund des Visusverlusts beim Ophthalmologen statt. Hier fällt auf, dass sich die Sehprobleme durch eine Brille nicht korrigieren lassen. In der ophthalmologischen Untersuchung können Makulaveränderungen in Form einer (unspezifischen) Schießscheibenmakulopathie auffallen. Zur weiterführenden ophthalmologischen Diagnostik gehören eine optische Kohärenztomographie (OCT) und ein Elektroretinogramm (ERG). In der OCT sucht man nach Zeichen für eine Störung der Photorezeptoren. Bei der juvenilen NCL sind die Potenziale im Elektroretinogramm schon früh nicht mehr nachweisbar.
Je nach Alter zum Zeitpunkt des Symptombeginns und aufgetretenen Symptomen kann die Diagnose auf verschiedene NCL-Formen eingegrenzt werden. Entsprechend unterscheidet sich die Diagnostik:
- Kongenitale Manifestation: Bei der kongenitalen Form (CLN10) sind, im Gegensatz zu allen anderen NCL-Formen, die Kinder bereits ab Geburt eindeutig krank. Die Kinder fallen mit Mikrozephalie und Krampfanfällen auf. Die Diagnostik erfolgt durch Messung der Aktivität von Cathepsin D in Leukozyten oder Hautfibroblasten. Das lysosomale Enzym wird durch das Gen CTSD kodiert. Bei eindeutig verminderter Aktivität steht die Diagnose fest, doch sollte zusätzlich eine Mutationsanalyse des betroffenen Gens durchgeführt werden, um der betroffenen Familie eine sichere genetische Beratung zu ermöglichen.
- Manifestation im Kleinkindalter: Hier sollten als erstes Enzymtests (Palmitoyl-Protein-Thioesterase auffällig bei CLN1 und Tripeptidyl-Peptidase-1 bei CLN2) durchgeführt werden. Sind diese unauffällig, so sollte in Gewebeproben nach Ceroid-Lipofuszinen gesucht werden. Alternativ kann eine Panel-Diagnostik der möglichen betroffenen Gene erfolgen, um eine genaue Diagnose zu stellen.
- Manifestation im Schulalter: Hier sollte ein Blutausstrich auf Lymphozytenvakuolen untersucht werden. Sind diese vorhanden, so gilt die Diagnose der juvenilen NCL (CLN3) als sicher. Es sollte zusätzlich eine molekulargenetische Analyse des CLN3-Gens erfolgen. Liegt dort keine Mutation vor, muss man an atypische Formen (z.B. protrahierter Verlauf einer CLN1 oder CLN2) oder andere Mutationen (z.B. CLN8 oder CLN9) denken. Zur Diagnosesicherung der CLN sollte auch hier in Gewebeproben nach den charakteristischen Ablagerungen gesucht werden.
- Manifestation im jungen Erwachsenenalter: Hier werden zunächst Enzymtests durchgeführt (Palmitoyl-Protein-Thioesterase, Tripeptidyl-Peptidase-1 oder Cathepsin D). Bei fehlender Enzymaktivität erfolgt eine molekulargenetische Analyse. Ist die Enzymaktivität unauffällig, werden auch hier Gewebeproben entnommen, um nach Ceroid-Lipofuszinen zu suchen.
Differentialdiagnostik
Aufgrund des ophthalmologischen Befundes wird teilweise zunächst die Diagnose einer Retinitis pigmentosa gestellt. Ihr Verlauf ist jedoch i.d.R. langsamer als bei der NCL.
Therapie
Für die meisten Formen der Erkrankung existieren keine kausalen Therapien und es sind rein symptomatische Behandlungen angezeigt. Dabei steht eine umfassende palliativmedizinische Betreuung im Vordergrund. Zur symptomatischen Therapie gehören u.a. eine antikonvulsive Therapie, eine neurologische, logopädische, ergo- und physiotherapeutische Anbindung sowie die psychologische Betreuung der Erkrankten und der Angehörigen. Weiterhin werden Symptome wie Schlafstörungen, Verhaltensauffälligkeiten, Schluckstörungen oder respiratorische Probleme behandelt.
Für die CLN2 besteht die Möglichkeit einer intraventrikulären Enzymersatztherapie mit Cerliponase alfa.[1]
Verschiedene Gentherapien befinden sich aktuell (2023) in der Entwicklung.
Weblinks
Forschungsaktivitäten werden unter anderem von der NCL-Stiftung unterstützt. Um eine frühzeitige Diagnose zu ermöglichen, werden Ärzte speziell für die NCL sensibilisiert, z.B. über Merkblätter.
- NCL-Stiftung – Für eine Zukunft ohne Kinderdemenz, abgerufen am 03.11.2023
- NCL-Stiftung – Merkblatt NCL (Kinderärzte), abgerufen am 03.11.2023
Literatur
- Simonati et al., Neuronal Ceroid Lipofuscinosis: The Multifaceted Approach to the Clinical Issues, an Overview, Frontiers in Neurology, 2022
- NCL-Stiftung – Neuronale Ceroid Lipofuszinosen (NCL, engl. Batten Disease), Stand des Wissens, 2022
- Bunesärztekammer – NCL-Kinderdemenz - Wenn Kinder ihr Fähigkeiten verlieren, Juni 2022
Quellen
- ↑ Pharmazeutische Zeitung Cerliponase alfa|Brineura®|40|2017], abgerufen am 29.11.2021