Apert-Syndrom





Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
Loslegennach Eugène Apert, Pädiater aus Paris (1868–1940)
Synonym: Akrozephalosyndaktylie-Syndrom
Englisch: Apert syndrome
Definition
Das Apert-Syndrom ist eine Erbkrankheit, die zu den Akrozephalosyndaktylie-Syndromen gehört und durch multiple Fehlbildungen charakterisiert ist.
- ICD-10: Q87.0
Ätiologie
Die Ursache des Apert-Syndroms ist eine Mutation des FGFR2-Gens auf Chromosom 10 am Genlokus 10q26. Das Gen kodiert für den Fibroblasten-Wachstumsfaktor-Rezeptor 2. In der überwiegenden Mehrzahl der Fälle liegt eine von zwei charakteristischen Punktmutationen in Exon 7 vor:[1]
- p.Ser252Trp (c.755C>G): im Genprodukt wird die Aminosäure Serin gegen Tryptophan ausgetauscht; diese Mutation tritt in etwa 2/3 aller Fälle auf und ist häufiger mit Gaumenspalte assoziiert
- p.Pro253Arg (c.758C>G): Austausch von Prolin gegen Arginin; tritt in etwa 1/3 der Fälle auf und ist häufiger mit schwereren Syndaktylie-Formen vergesellschaftet
Die Vererbung erfolgt autosomal-dominant. Es besteht eine vollständige Penetranz, das heißt, jeder Träger des mutierten Gens ist auch von der Erkrankung betroffen. Die Expressivität des Apert-Syndroms ist allerdings variabel, d.h. die Symptome sind bei den Betroffenen unterschiedlich stark ausgeprägt.
Häufig sind De-novo-Mutationen Ursache des Apert-Syndroms. Dabei spielt das Alter des Vaters eine wichtige Rolle. Das Apert-Syndrom tritt häufiger bei Kindern älterer Väter auf, da mit steigendem väterlichem Alter die Akkumulation von Neumutationen in den väterlichen Keimzellen zunimmt.[2] Etwa eines von 130.000 Neugeborenen ist von der Erkrankung betroffen.
Klinik
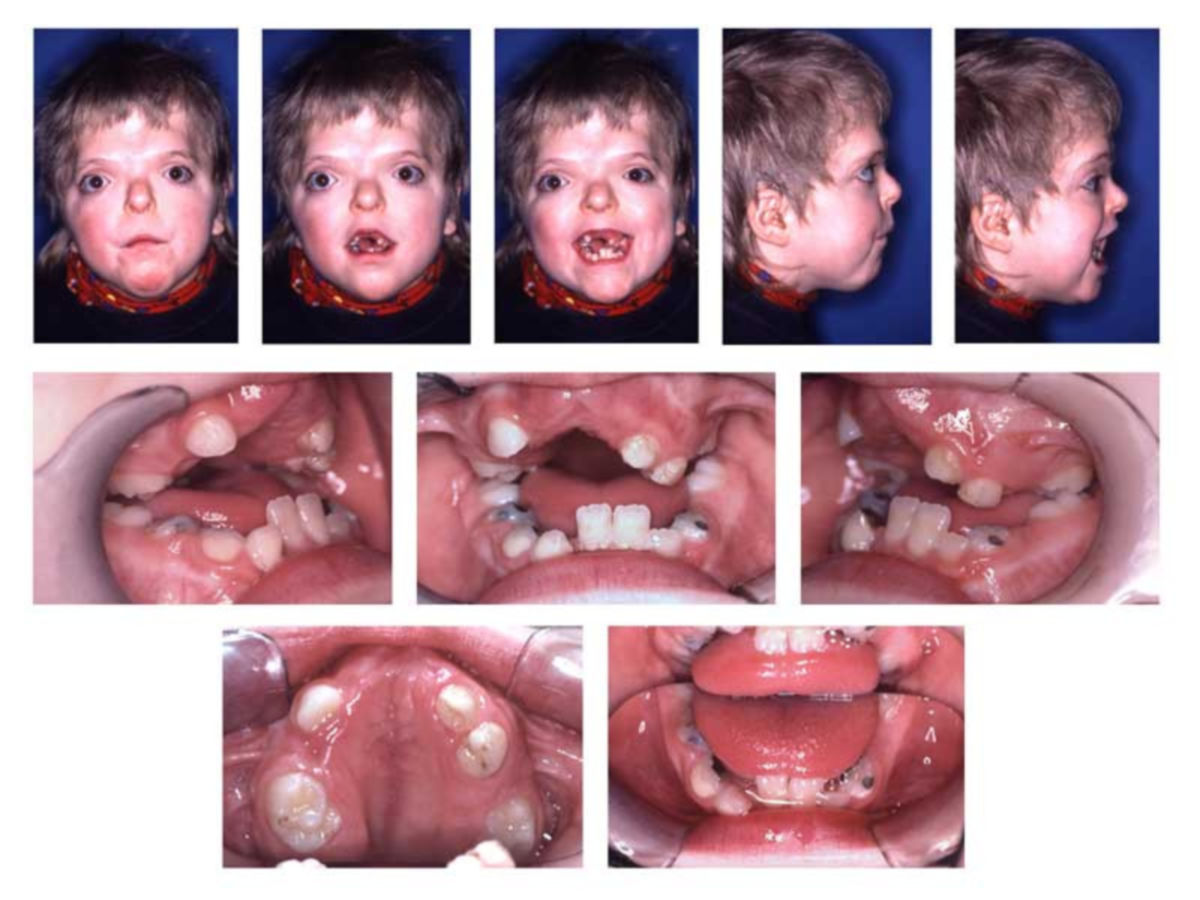
Folgende Symptome treten häufig im Rahmen des Apert-Syndroms auf:
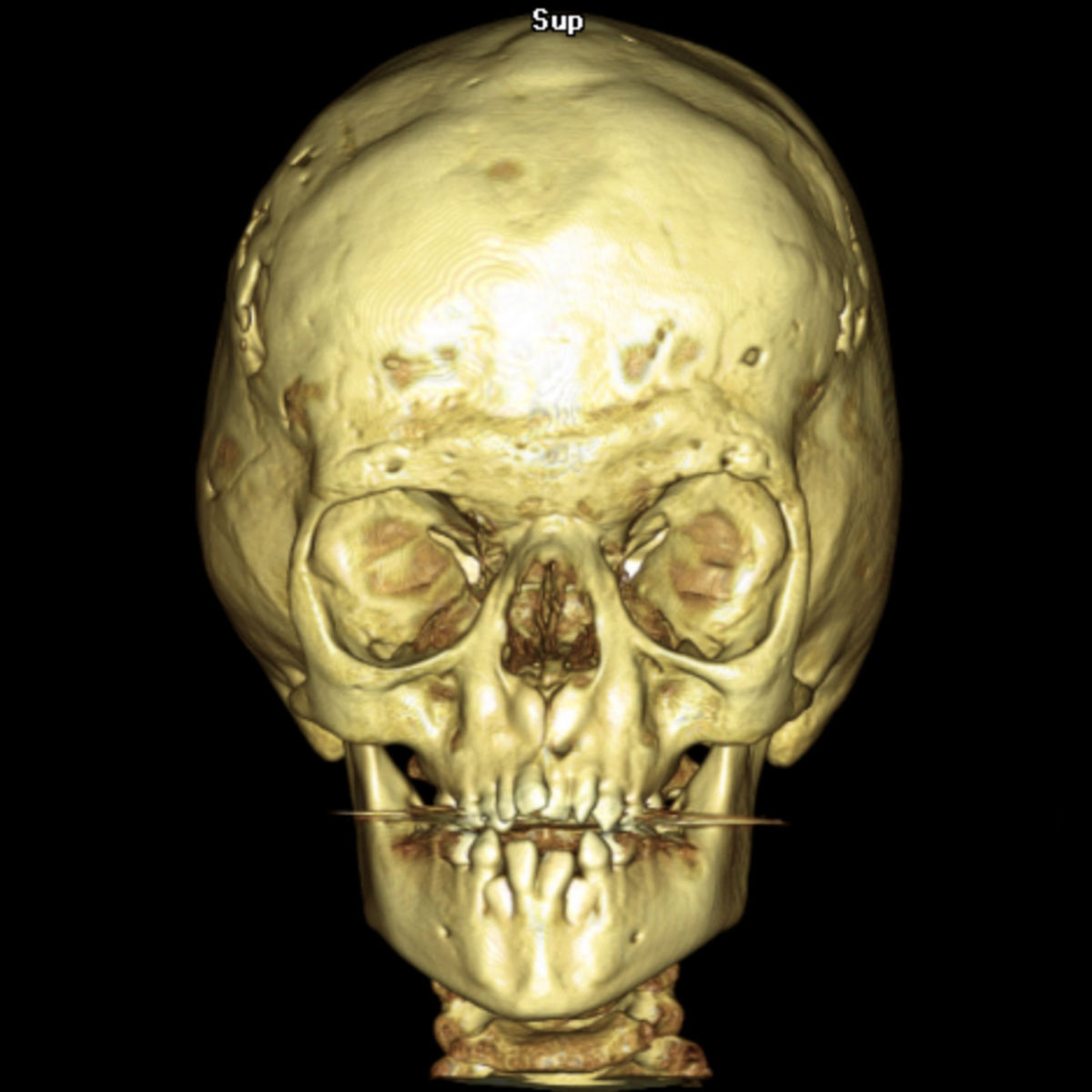
- Fehlbildungen des Kopfes
- Turmschädel durch vorzeitige Verknöcherung der Koronarnähte des Schädels (Kraniosynostose)
- Fehlbildungen des Gesichts
- flache Orbita
- Exophthalmus
- Hypertelorismus
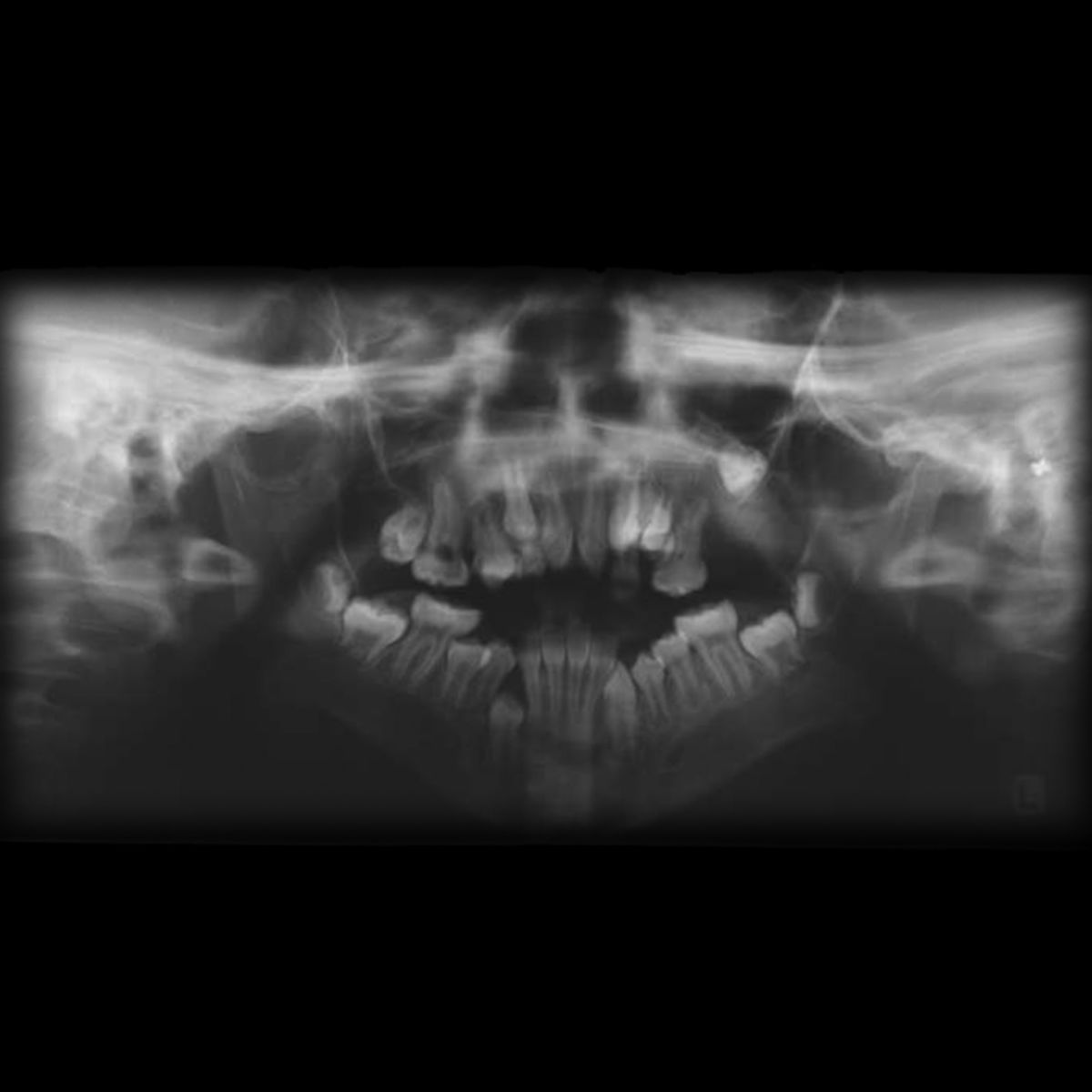
- Fehlbildungen des Oberkiefers
- Gaumenspalte
- pseudomandibuläre Prognathie
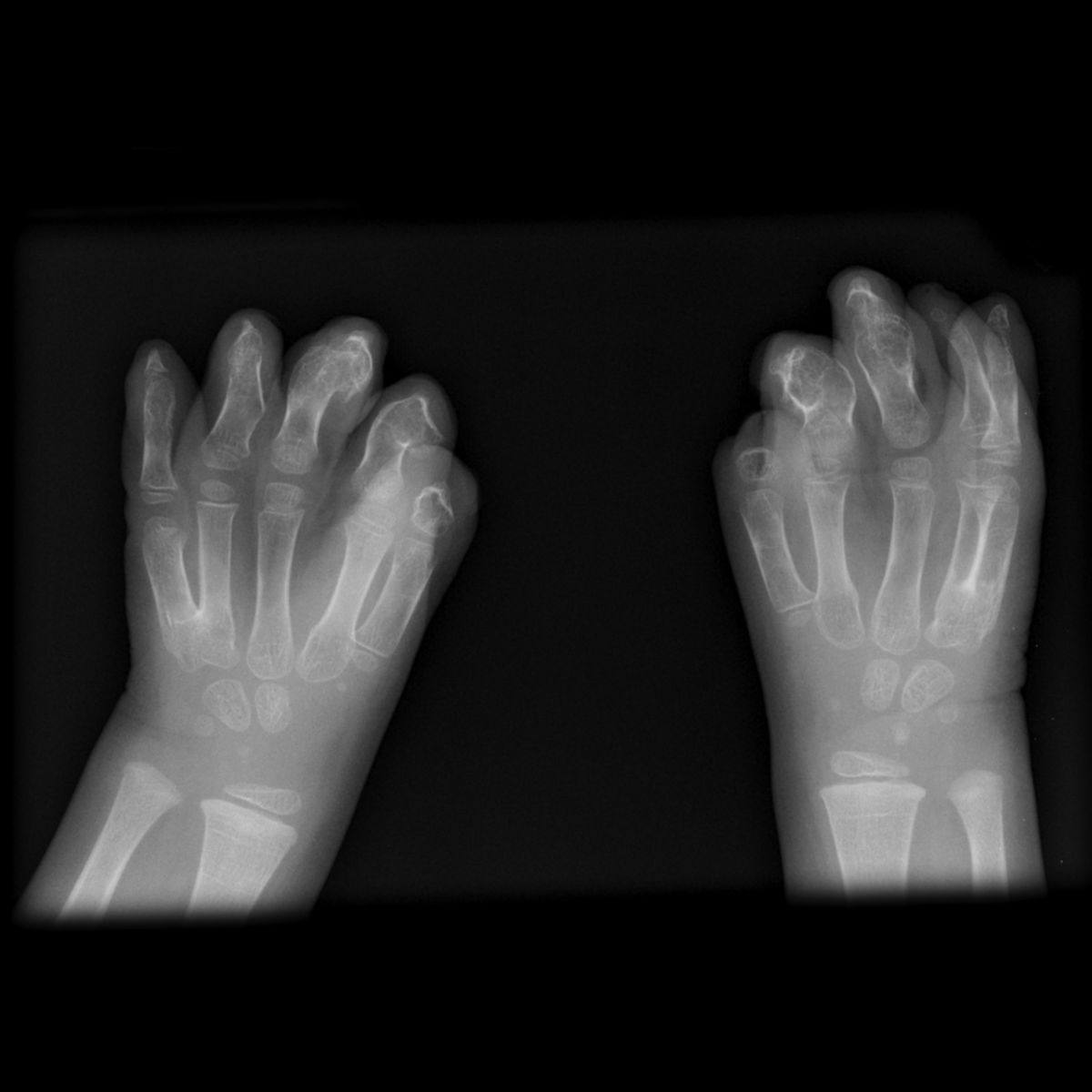
- Fehlbildungen der Hände und Füße
- Syndaktylie (immer symmetrisch, oft mit gemeinsamen Fingernägel)
- kurzer Daumen mit radialer Deviation
- Versteifung der Mittelgelenke der Finger (z.T. fehlen diese vollständig)
- Löffelhände
- Synostosen
- Symphalangie
- Brachydaktylie
- Anomalien der Schulter- und Ellenbogengelenke (werden häufig übersehen, sollten aber gezielt untersucht werden)[3]
- dentale Anomalien
- Zahnagenesie (ca. 37 % d.F., häufig bilaterales Fehlen der mandibulären zweiten Prämolaren)[4]
- Skoliose
- Schwerhörigkeit
- Fehlsichtigkeit
- Atemwegsobstruktion (häufig mit obstruktiver Schlafapnoe)
- Intelligenzminderung (in ca. 50–80 % der Fälle; Ausprägung variabel)[5]
- Ventrikulomegalie (bei etwa 40 %)[6]
- Hydrozephalus (ca. 2–5 %)[7]
Die Handfehlbildungen werden in drei Typen differenziert, wobei Typ I die leichteste Form der Deformation darstellt, Typ III die schwerste.[8]
Komplikationen
- Erhöhter Hirndruck durch Kraniosynostose, insbesondere bei verzögerter chirurgischer Dekompression
- Optikusatrophie und Papillenödem als Folge eines chronisch erhöhten intrakraniellen Drucks; eine bilaterale Beteiligung des Sehnervs ist häufig[9]
- rezidivierende Otitis media durch Fehlbildungen der Gehörknöchelchen und des Mittelohrs
- Sehverschlechterung bis zur Erblindung bei schwerem Exophthalmus
- psychische Folgeerkrankungen
Differentialdiagnosen
Das Apert-Syndrom wird zu den kraniofazialen Fehlbildungen gerechnet. Zu dieser Gruppe gehören außerdem folgende Erkrankungen, die differentialdiagnostisch abzugrenzen sind:
- Crouzon-Syndrom: Kraniosynostose ohne Syndaktylie
- Pfeiffer-Syndrom: Kraniosynostose mit breiten Daumen und Großzehen, meist mildere Syndaktylie als beim Apert-Syndrom
- Saethre-Chotzen-Syndrom: Kraniosynostose mit Ptosis und milder Syndaktylie
- Carpenter-Syndrom: Kraniosynostose mit Polydaktylie und Adipositas
Diagnostik
Das Apert-Syndrom kann schon vor der Geburt im Rahmen der Pränataldiagnostik auffallen. Im Feinultraschall (ab ca. 3. Schwangerschaftsmonat) sind typische Merkmale erkennbar, darunter eine Turrizephalie infolge bikoronarer Nahtsynostose, Mittelgesichtshypoplasie sowie das charakteristische Syndaktylenmuster der Hände und Füße. Die fetale MRT kann ergänzend intrakranielle Begleitfehlbildungen identifizieren.[10] Die Diagnose erfolgt dann meist durch Chorionzottenbiopsie (ab der 10. Schwangerschaftswoche) oder Fruchtwasseruntersuchung (ab der 15. Schwangerschaftswoche) mittels gezielter FGFR2-Sequenzierung.
Nach der Geburt geben die typischen Symptome Hinweis auf die Erkrankung. Die Bestätigung der Diagnose ist durch molekulargenetische Methoden möglich.
Bei Diagnosestellung sollte eine humangenetische Beratung der Eltern erfolgen, insbesondere bei fortgeschrittenem Vateralter oder positivem Familienbefund.
Therapie
Zur Behandlung der Schädel-Deformität ist oftmals eine chirurgische Nahtöffnung der Schädelnähte notwendig, damit die Gehirnentwicklung nicht beeinträchtigt wird. Der Eingriff erfolgt typischerweise innerhalb der ersten 6–12 Lebensmonate. In ausgewählten Fällen stehen auch minimalinvasive endoskopische Verfahren zur Verfügung.[11] Die Mittelgesichtsanomalien werden durch eine Distraktionsosteogenese behandelt.
Außerdem wird die Syndaktylie chirurgisch getrennt und danach eine Stellungskorrektur der Finger und Zehen vorgenommen. Die Eingriffe richten sich dabei nach dem genauen Typ der Fehlbildung. Neuere Techniken ermöglichen in ausgewählten Fällen die Syndaktylietrennung ohne Hauttransplantation durch kontrollierte Weichteildistraktion mittels externem Fixateur.[12]
Manchmal muss auch das Mittelohr operiert werden, weil Gehörknöchelchen beim Apert-Syndrom z.T. nicht ausgebildet sind.
Die psychische Belastung durch soziale Ablehnung und zahlreiche Operationen kann bei betroffenen Kindern zu psychischen Problemen führen, weshalb auch eine psychosoziale Betreuung Teil der Therapie sein sollte.
Prognose
Die Lebenserwartung beim Apert-Syndrom ist bei adäquater chirurgischer und multidisziplinärer Versorgung nicht grundsätzlich verkürzt. Die kognitive Entwicklung ist variabel. Während ein Teil der Betroffenen eine Intelligenzminderung aufweist, erreichen andere eine normale oder grenzwertige Intelligenz. Die Prognose hängt wesentlich vom Ausmaß des Hirndrucks in der frühen Kindheit, dem Zeitpunkt der kranialen Dekompression sowie von der Intensität der frühzeitigen Förderung ab.
Quellen
- ↑ Brajadenta et al., Molecular analysis of exon 7 of the fibroblast growth factor receptor 2 (FGFR2) gene in an Indonesian patient with Apert syndrome: a case report, J Med Case Rep, 2019
- ↑ Aitken, Male reproductive ageing: a radical road to ruin, Hum Reprod, 2023
- ↑ Khabyeh-Hasbani et al., Contemporary Management of the Upper Limb in Apert Syndrome: A Review, Plast Reconstr Surg Glob Open, 2024
- ↑ Becerril Santos et al., Prevalence and Patterns of Permanent Tooth Agenesis in Patients With Crouzon or Apert Syndrome: A Systematic Review and Meta-Analysis, Orthod Craniofac Res, 2026
- ↑ Maliepaard et al., Intellectual, behavioral, and emotional functioning in children with syndromic craniosynostosis, Pediatrics, 2014
- ↑ Jönsson et al., Prevalence and treatment outcomes of hydrocephalus among children with craniofacial syndromes, J Plast Surg Hand Surg, 2025
- ↑ Collmann et al., Hydrocephalus in craniosynostosis: a review, Childs Nerv Syst, 2005
- ↑ Upton et al., Resection of Fourth-to-Fifth Metacarpal Synostosis and Fascial Interposition for Creation of a Functional Grip/Pinch in the Apert Hand, Plast Reconstr Surg, 2025
- ↑ Manara et al., Cranial nerves involvement in craniosynostosis: a systematic review, Childs Nerv Syst, 2026
- ↑ Li et al., From FGFR2 mutations to precision management: a review of prenatal diagnosis and multidisciplinary interventions in Apert syndrome, Front Pediatr, 2025
- ↑ S2k-Leitlinie Diagnostik und Therapie von Patienten mit Kraniosynostosen, abgerufen am 18.05.2026
- ↑ Kinoda et al., Webplasty using an external fixator for complex syndactyly caused by Apert syndrome, J Orthop Sci, 2025