Morbus Hirschsprung










Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
Loslegennach dem dänischen Pädiater Harald Hirschsprung (1830-1916)
Synonyme: kongenitales Megakolon, aganglionäres Megakolon, Megacolon congenitum, intestinale Aganglionose
Englisch: congenital megacolon, Hirschsprung’s disease
Definition
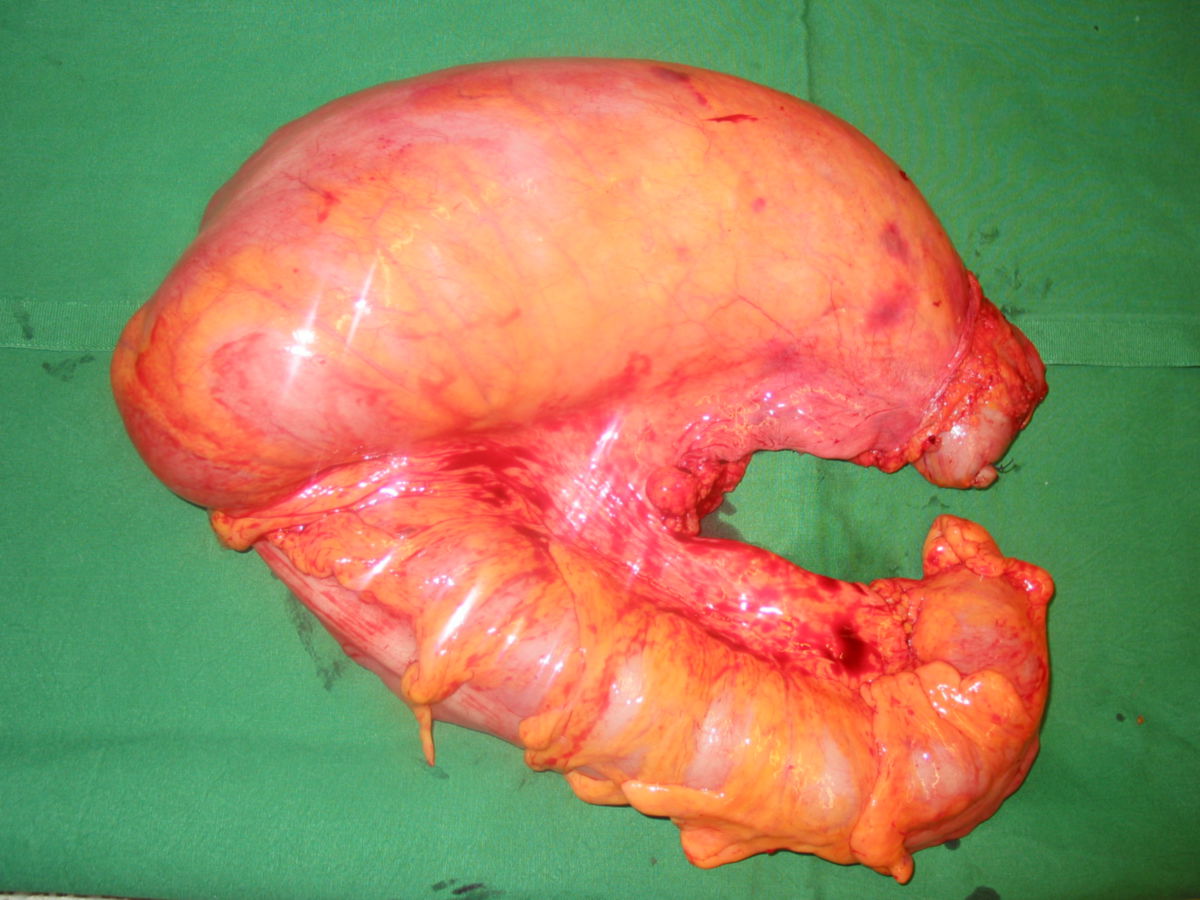
Der Morbus Hirschsprung ist eine angeborene Veränderung der neuronalen Strukturen des enterischen Nervensystems. Durch das segmentale Fehlen der Ganglienzellen im Plexus myentericus und Plexus submucosus (Aganglionose) kommt es zu einer tonischen Kontraktion des betroffenen Darmabschnittes. Dieses Passagehindernis führt zur Dilatation der prästenotischen Dickdarmareale (Megakolon).
Epidemiologie
Die Häufigkeit dieser Erkrankung beträgt etwa 1/5.000. Männliche Säuglinge sind dreimal häufiger betroffen. In 80 % der Fälle ist die Aganglionose auf das Rektum und das Sigma beschränkt. Bei 8 % der Patienten ist der komplette Dickdarm betroffen, sehr selten findet sich die Aganglionose auch im Dünndarm.
Ätiologie
Die genaue Ursache der angeborenen Aganglionose ist derzeit (2026) unklar. Vermutlich liegt der Erkrankung eine unvollständige Besiedlung der Darmwand mit Nervenzellvorläufern aus der Neuralleiste zugrunde.
Mutationen u.a. von RET, Endothelin-3, Endothelin-B-Rezeptor, Endothelin-konvertierendem Enzym 1, Sox10 und SMADIP1 sind beschrieben. Bei weniger als 10 % der betroffenen Patienten kann eine verantwortliche Mutation nachgewiesen werden. Die Erkrankung tritt in 20 % d.F. familiär gehäuft, in 80 % d.F. sporadisch auf. Die Vererbung kann autosomal-dominant oder -rezessiv sein.
Des Weiteren tritt der Morbus Hirschsprung in 10 % d.F. im Rahmen einer Trisomie 21 auf. Weitere syndromale Formen sind:
- Waardenburg-Shah-Syndrom (zusätzlich Pigment- und Hörstörungen)
- Mowat-Wilson-Syndrom (zusätzlich kognitive und motorische Retardierung mit Epilepsie)
- Haddad-Syndrom (zusätzlich Undine-Syndrom)
Pathophysiologie
Aufgrund des Fehlens von enterischen Neuronen im betroffenen Darmsegment werden keine relaxierenden Neurotransmitter ausgeschüttet. Die tonische Kontraktion führt zu einer funktionellen Obstruktion. Der Aufstau des Stuhles bedingt in den proximalen Anteilen eine massiven Dilatation (Megakolon).
Symptomatik
Die betroffenen Neugeborenen zeichnen sich meist durch ein immens aufgetriebenes Abdomen aus. Das Mekonium wird verspätet abgegeben, manchmal hat sich bereits ein Ileus entwickelt.
Allerdings kann sich ein Morbus Hirschsprung auch erst bei der Umstellung auf Breikost rund um den fünften Lebensmonat zeigen. Das Kind verweigert dann die Nahrungsaufnahme und setzt kaum noch Stuhl ab. Während bei der ausschließlichen Ernährung mit Muttermilch der Stuhl weich ist und sich entsprechend leichter durch die Stenose schieben kann, dickt der Stuhl nach der Nahrungsumstellung durch die Ballaststoffe entsprechend ein. Der Stuhlverhalt kann meist schon palpatorisch festgestellt werden.
Wird eine frühzeitige artifizielle Stuhlausleitung versäumt, kann sich eine schwere nekrotisierende Enterokolitis mit nachfolgender Sepsis entwickeln. Bei der rektalen Untersuchung, fällt der typisch erhöhte Sphinktertonus auf, bei leerer Rektumampulle und stuhlgefülltem Abdomen.
Beschränkt sich die Aganglionose nur auf ein kurzes Darmsegment, so werden Kinder oftmals erst nach dem Abstillen auffällig. In seltenen, milden Fällen manifestiert sich ein Morbus Hirschsprung erst im Erwachsenenalter.
Diagnostik
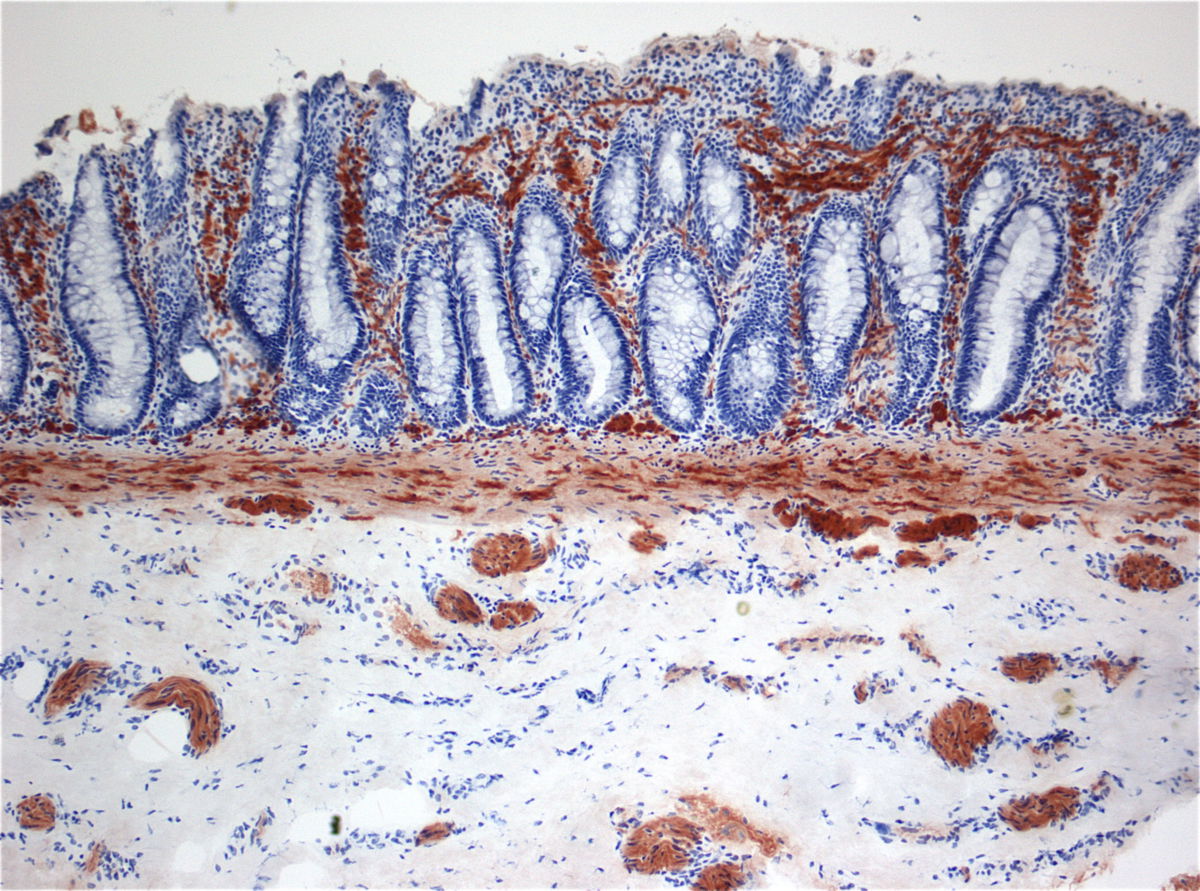
Standarddiagnostik bei Verdacht auf Morbus Hirschsprung ist die Etagenbiopsie des Rektums mittels Saugbiopsie. Der histopathologische Befund der Schleimhautbiopsie ist gekennzeichnet durch:
- Fehlen intramuskulärer Ganglienzellen
- erhöhte Acetylcholinesterase-Konzentration im enzymhistochemischen Färbepräparat
- Hyperplasie cholinerger Nervenfasern
Weitere diagnostische Methoden umfassen:
- anorektale Manometrie: Nach Dehnung der rektalen Schleimhaut fehlt der Relaxationreflex des Sphincter internus.
- Kolon-Kontrastdarstellungen: In der Röntgenaufnahme bei Kolonkontrasteinlauf ohne vorheriges Abführen fällt der charakteristische Lumensprung des Darmes auf – das prästenotische, dilatierte Megakolon und das distale, engestellte, aganglionäre Segment.
- molekulargenetische Untersuchung (MEN2A-, MTC-Mutationen, RET-Screening)
In den seltenen Fällen, in denen ein Morbus Hirschsprung erst im Erwachsenenalter diagnostiziert wird, kommt initial die anorektale Manometrie vor eventueller Durchführung einer tiefen Rektumbiopsie zum Einsatz.
Therapie
Um lebensbedrohliche Komplikationen wie Sepsis und Durchwanderungsperitonitis zu verhindern, muss eine frühzeitige operative Therapie angestrebt werden. Initial erfolgt in der Regel eine konservative Entlastung des Darms durch rektale Spülungen. Eine primäre Anlage eines Anus praeter ist nicht obligat, sondern ausgewählten Fällen (z.B. bei schwerer Enterokolitis, Perforation oder ausgeprägter Dilatation) vorbehalten.
Die definitive Therapie besteht in einer Durchzugsoperation (Pull-through-Operation) mit Resektion des aganglionären Darmsegments und Anastomosierung des normoganglionären Darms an den Analkanal. Je nach Befund und Zentrum kommen z.B. Verfahren nach Swenson, Soave oder Duhamel sowie transanale endorektale Techniken (z.B. endorektale transanale Rektosigmoidektomie nach de la Tolle), ggf. laparoskopisch assistiert, zum Einsatz. Die beiden Resektionsränder werden im Anschluss in der Regel als koloanale Anastomose vereinigt. Abhängig von der Technik kann dabei ein Stapler eingesetzt werden.
Durch eine etwa 1–2 cm breite Sphinkteromyotomie kann die Kontraktilität des Musculus sphincter ani internus vermindert werden, sie stellt jedoch kein Standardverfahren der Primärtherapie dar und ist heute (2026) speziellen funktionellen Problemkonstellationen vorbehalten.
siehe auch: Jirásek-Zuelzer-Wilson-Syndrom, Toxisches Megakolon
Literatur
- Fitze et al.: S1-Leitlinie Morbus Hirschsprung des Rektosigmoids, 2023, zuletzt abgerufen am 17.02.2026
- Universitätsklinikum Leipzig, Klinik und Poliklinik für Kinderchirurgie: Morbus Hirschsprung, zuletzt abgerufen am 17.02.2026