Erythropoetische Protoporphyrie



Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenSynonyme: Protoporphyrinämische Lichtdermatose, Protoporphyria erythropoetica
Definition
Die erythropoetische Protoporphyrie, kurz EPP, ist eine seltene, familiäre Erkrankung aus der Gruppe der Photodermatosen, die durch urtikarielle und pachydermieartige Hautveränderungen imponiert, die auf einem Defekt der Ferrochelatase beruhen.
Epidemiologie
Die protoporphyrinämische Lichtdermatose ist eine seltene Erkrankung, die bei allen Völkern auftritt. Sie wird unregelmäßig autosomal-dominant vererbt, wobei Männer doppelt so häufig betroffen sind wie Frauen. Die Inzidenz beträgt in Deutschland etwa 1:100.000.
Ätiopathogenese
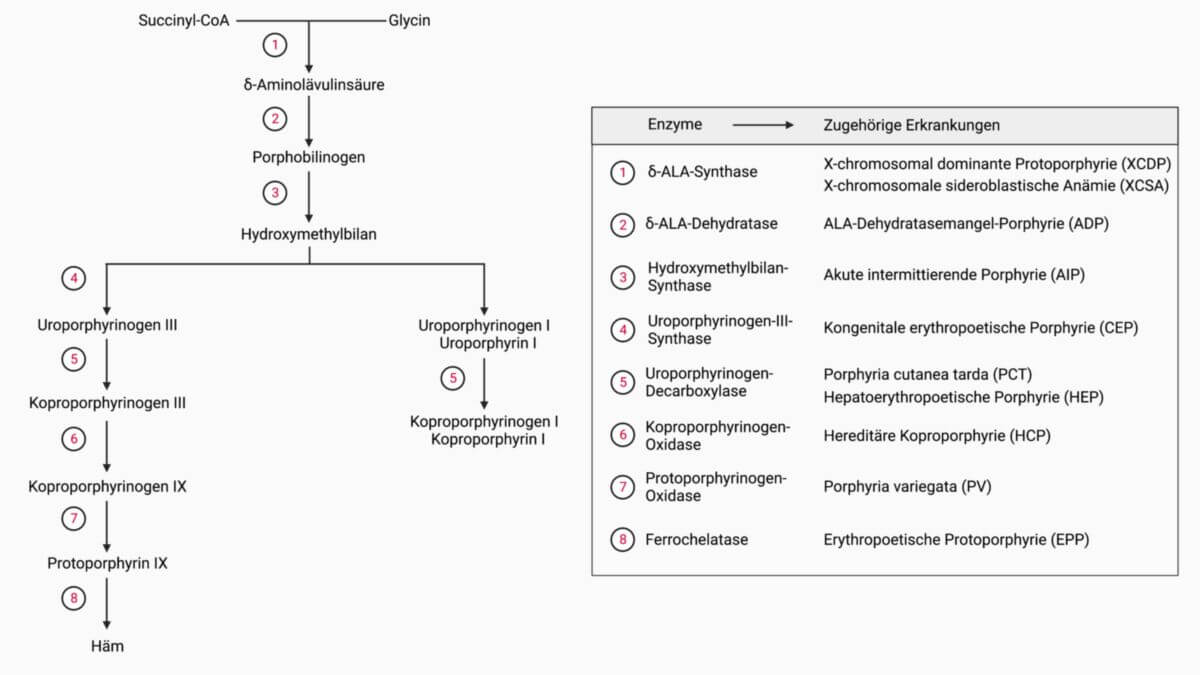
Die Erkrankung beruht auf dem kongenitalen Defekt der Ferrochelatase, welcher zu einer ausgeprägten Anreicherung von Protoporphyrin in den Erythrozyten führt. In lichtexponierten Hautarealen kommt es zur Photohämolyse und dem Austreten von Protoporphyrinen ins perivaskuläre Gewebe. Die durch den Austritt von Protoporphyrinen ausgelösten Zellschäden führen zu den typischen klinischen Symptomen.
Klinik
Bei betroffenen Patienten führt Sonnenexposition, vor allem im Bereich des Handrückens und des Gesichts, insbesondere an den Wangen, der Nase und dem Kinn, zu einer Rötung der Haut, die stark brennt und juckt und nach wenigen Stunden von einem flüchtigen, urtikariellen oder von einem derben, über Tage persistierendem Infiltrat gefolgt wird. An der aufgetriebenen Haut sind Einblutungen, seltener Blasenbildungen möglich.
Das Aufkratzen der betroffenen Areale kann zu diskreten Narben führen. Wiederholte Sonnenlichtexposition führt zu einer Verstärkung der Symptome, so dass es schließlich durch eine perivaskuläre Einlagerung von Lipoproteinen zu einer Pachydermie kommt.
Durch die Einlagerung von Protoporphyrinen in die Leber treten bei einem Teil der Patienten Leberschäden auf, die in eine Leberzirrhose münden können. Desweiteren kommt es zur Bildung protoporphyrinhaltiger Gallensteine.
Diagnose
Die Diagnose kann anhand der Klinik vermutet werden und wird durch die Protoporphyrin-Fluoreszenz bestätigt. Diese zeigt eine Fluoreszenz von etwa 10% der Erythrozyten in der verwendenten Erythrozytensuspension, bevor diese durch Photohämolyse zersetzt werden. Im Stuhl werden vermehrt Protoporphyrine ausgeschieden, während der Gehalt an Porphyrinen im Urin normal ist.
Differenzialdiagnose
Differenzialdiagnostisch sollte an die kongenitale erythropoetische Porphyrie, an phototoxische Arzneimittelreaktionen, Kontaktdermatitis, Angioödem sowie an eine Bleivergiftung gedacht werden.
Therapie
Konstanter und hoher Lichtschutz sowie die Vermeidung von direkter Sonnenlichtexposition sind obligat. In der Vergangenheit wurde häufig beta-Karotin als oraler Lichtschutz verwendet, welcher zu einer leichten Gelbfärbung der Haut führte. Diese Methode wird heute nicht mehr empfohlen, da sie nicht durch kontrollierte klinische Studien überprüft wurde und somit nicht den Anforderungen der evidenzbasierten Medizin entspricht.
Ein zusätzlicher Behandlungsansatz ist die Therapie mit Afamelanotid, einem Wirkstoff aus der Gruppe der Melanocortin-Rezeptor-Agonisten. Afamelanotid fördert auf hormonellem Weg die Bildung von Melanin und verstärkt so die Hautpigmentierung ohne UV-Exposition. Der Wirkstoff wird in Form eines subkutanen Implantats verabreicht, das etwa zwei Monate wirksam ist.
Bei Patienten, die Sonnenlicht meiden müssen, sollte an einen Vitamin-D-Mangel gedacht werden.
Prognose
Unter konsequentem Lichtschutz ist die Prognose gut, jedoch kann die Lebensqualität der betroffenen Patienten durch die Vermeidung von Sonnenlichtexposition mehr oder weniger stark eingeschränkt sein. Durch Gabe von Afamelanotid können diese Einschränkungen teils vermieden werden.
Literatur
- European Porphyria Initiative – The Porphyrias, abgerufen am 13.06.2023
- orpha.net – Protoporphyrie, erythropoetische, autosomale Form, abgerufen am 13.06.2023
- MSDManual – Erythropoetische Protoporphyrie und X-linked Protoporphyrie, abgerufen am 13.06.2023