Kongenitale erythropoetische Porphyrie






Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenSynonyme: Morbus Günther, erythropoetische Porphyrie
Englisch: congenital erythropoietic porphyria, CEP
Definition
Unter der kongenitalen erythropoetischen Porphyrie, kurz CEP, versteht man eine seltene, schwere Erkrankung, die mit Photosensibilität, Narben sowie Narbenkarzinomen einhergeht.
Epidemiologie
Es handelt sich um eine sehr seltene, schwere Stoffwechselerkrankung.
Ätiopathogenese
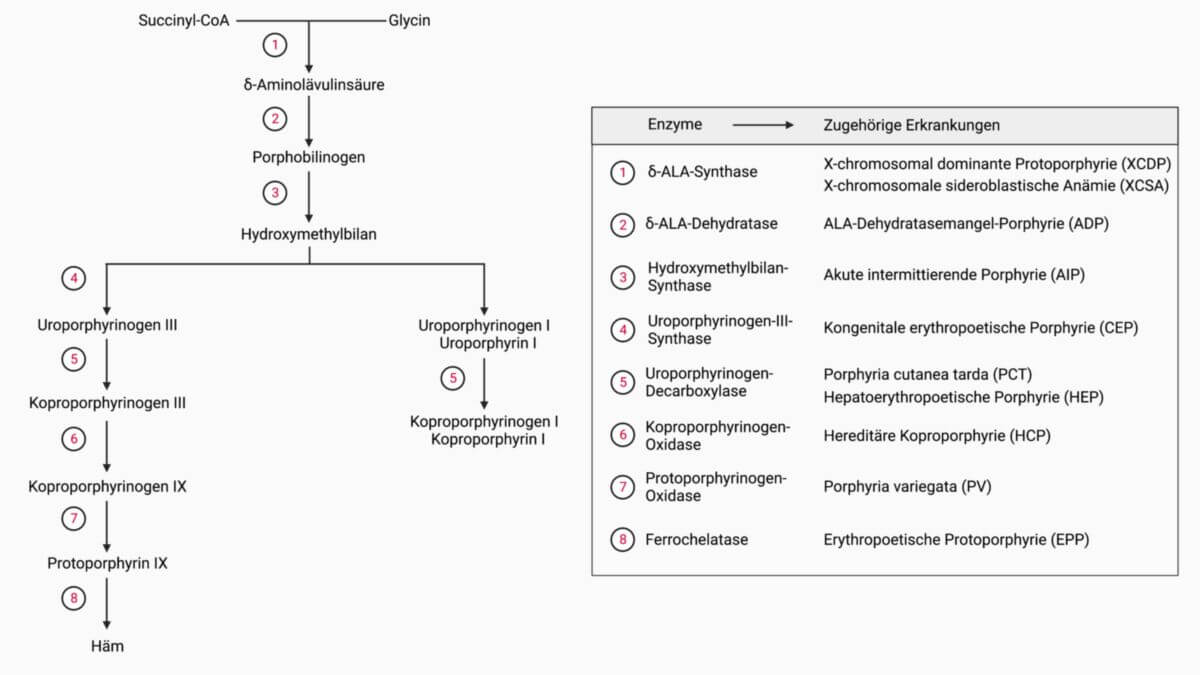
Die Erkrankung beruht auf einem Defekt der Uroporphyrinogen-III-Synthase auf dem Chromosom 10, der autosomal-rezessiv vererbt wird und zu einer Erhöhung der Gesamtporphyrine in den Erythrozyten führt. Es sind zahlreiche Mutationen bekannt. Je nach vorliegender Mutation kann eine Restaktivität des Enzyms bestehen, sodass die Symptomschwere sich von Fall zu Fall unterscheidet. Die Eltern der betroffenen Patienten sind häufig blutsverwandt.
Klinik
Charakteristisch ist das Auftreten von Hautrötungen nach Lichtexposition, die von hämorrhagischen Bläschen und Blasen gefolgt werden. Aus diesen Bläschen und Blasen gehen Erosionen und Ulzerationen hervor, die nur schlecht abheilen und häufig superinfiziert sind.
Durch wiederholte Lichtexpositionen an den Akren kommt es zu Mutilationen mit sklerodermieartigem, derbem Charakter sowie Hypo- oder Hyperpigmentierung.
Am Kopf ist das Auftreten einer fleckförmigen bis flächigen, narbigen Alopezie möglich. Typischerweise kommt es zusätzlich zu einer Hypertrichose der lichtexponierten Hautareale, insbesondere am Handrücken, den Wangen und den Vorderarmen.
Eine Splenomegalie sowie eine hämolytische Anämie sind ebenfalls häufig. An den mutilierten Hautarealen können Narbenkarzinome entstehen.
Diagnose
Zur Diagnose führen Klinik und typische Untersuchungsbefunde. Der Urin der betroffenen Patienten ist rosarot gefärbt. Die Zähne und Knochen können ebenfalls eine rötliche Verfärbung (Erythrodontie) annehmen, die unter Wood-Licht intensiviert wird.
In den Erythrozyten ist die Gesamtmenge an Porphyrinen erhöht; im Urin und Stuhl werden große Mengen and Porphyrinen ausgeschieden, wobei sechzig bis neunzig Prozent der ausgeschiedenen Porphyrine der Isomeren-Reihe I und nicht der Reihe III angehören.
Histopathologisch zeigen sich subepidermale Blasen, wobei die Veränderungen jedoch nicht richtungsweisend sind.
Die Diagnose wird durch eine molekulargenetische Untersuchung gesichert.
Differenzialdiagnose
Differenzialdiagnostisch sollte an andere Formen der Porphyrie gedacht werden sowie an die Xeroderma pigmentosum und an Poikilodermien.
Therapie
Eine ursächliche Therapie ist zur Zeit (2023) nicht verfügbar. Strenger Lichtschutz sowie das strikte Meiden jeglicher Sonnenlichtexposition sind obligat. In leichteren Fällen kann die orale Einnahme von beta-Carotin eine Linderung der Symptome bringen, wobei die Behandlung schon im Kindesalter begonnen werden sollte. In schweren Verlaufsfällen kann ggf. eine allogene Stammzelltransplantation erfolgen.
Gentherapien und andere kausal ausgerichtete Behandlungsformen befinden sich in klinischer Entwicklung.
Prognose
Die Lebensqualität der betroffenen Patienten ist deutlich vermindert und die Lebenserwartung durch die hämolytische Anämie sowie die Narbenkarzinome eingeschränkt.