Adenoid-zystisches Speicheldrüsenkarzinom











Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenSynonym: Zylindrom (veraltet)
Englisch: adenoid cystic carcinoma
Definition
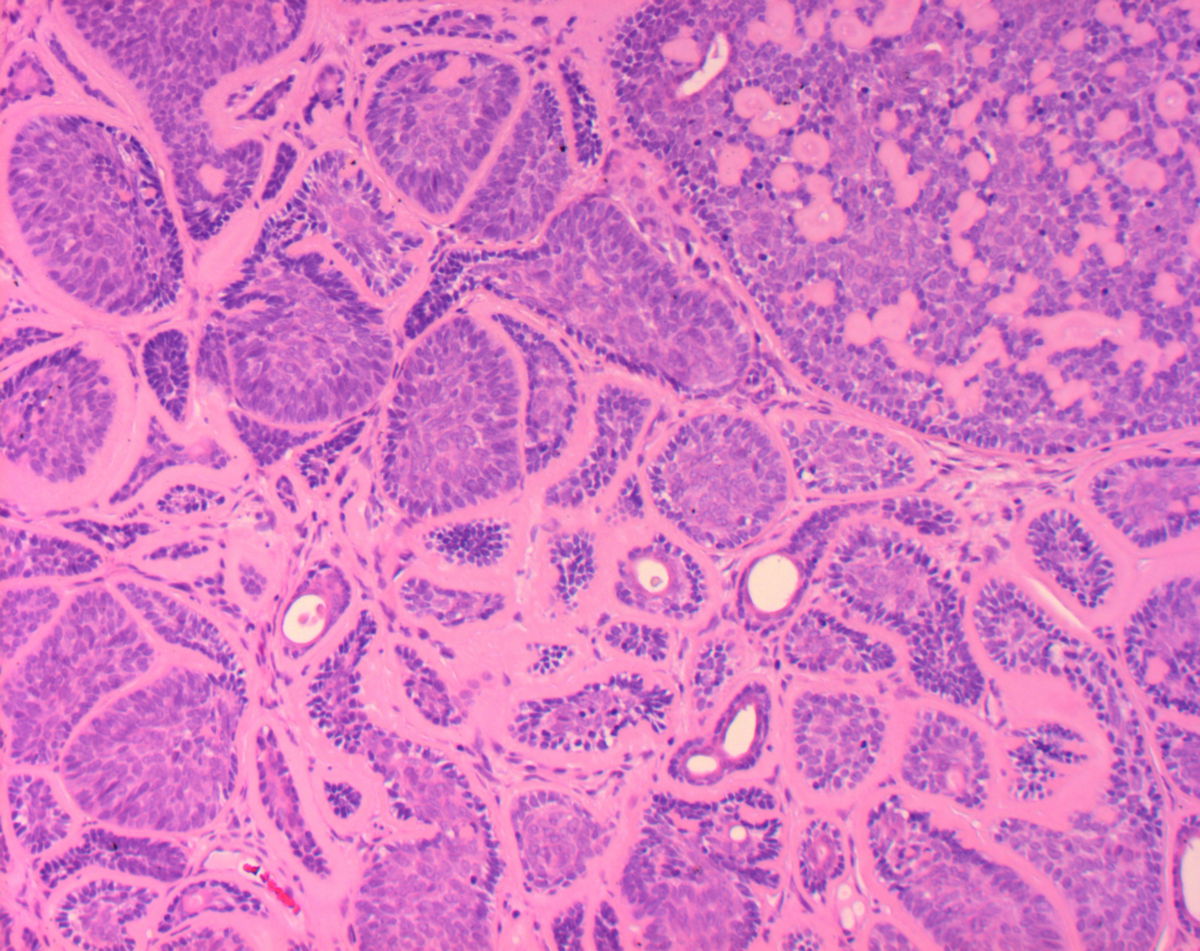
Das adenoid-zystische Speicheldrüsenkarzinom ist ein maligner epithelialer Tumor der Speicheldrüsen, der durch ein charakteristisches kribriformes, tubuläres oder solides Wachstumsmuster sowie eine ausgeprägte Perineuralscheideninvasion gekennzeichnet ist.
Epidemiologie
Das adenoid-zystische Karzinom macht etwa 5 bis 10 % der Speicheldrüsentumoren aus. Es tritt bevorzugt in den kleinen Speicheldrüsen, insbesondere am harten Gaumen, auf und ist in der Glandula parotidea vergleichsweise selten. Weitere Lokalisationen sind die Glandula submandibularis, die Glandula sublingualis, die Zunge sowie die Nasennebenhöhlen. Außerhalb des Kopf-Hals-Bereichs kommen adenoid-zystische Karzinome auch in Haut, Lunge, Mamma und weiblichem Genitaltrakt vor.[1]
Der Altersgipfel liegt zwischen dem 4. und 6. Lebensjahrzehnt. Ein Auftreten im Kindesalter ist selten. Frauen sind etwas häufiger betroffen als Männer.
Histologie
Der Tumor imponiert als unscharf begrenzte, derbe Raumforderung mit infiltrativem Wachstum. Histologisch werden drei Wachstumsmuster unterschieden, die häufig nebeneinander vorliegen:
- kribriformes Muster (siebartig, klassisch)
- tubuläres Muster
- solides Muster
Weitere Charakteristika sind:
- biphasische Zellpopulation aus duktalen und myoepithelialen Zellen
- zystenartige Pseudolumina mit PAS-positivem, basalmembranartigem Material
- meist niedrige Mitoserate
- ausgeprägte perineurale und perivaskuläre Ausbreitung
Ein hoher Anteil solider Tumoranteile (> 30 %) gilt als prognostisch ungünstig.
Molekularpathologie
Charakteristisch ist eine Translokation t(6;9)(q22–23;p23–24) mit Bildung eines MYB-NFIB- bzw. seltener MYBL1-NFIB-Fusionsgens, das in einem Großteil der Tumoren nachweisbar ist.[2][3]
Klinik
Die Symptomatik ist abhängig von Lokalisation und Ausdehnung:
- langsam wachsende, häufig schmerzhafte Schwellung
- diffuse Schmerzen und Parästhesien durch perineurale Infiltration
- Fazialisparese bei Befall der Glandula parotidea
- zervikale Lymphadenopathie
- bei palatinaler Lokalisation Ulzerationen der Schleimhaut
Diagnostik
Die Diagnostik umfasst klinische, bildgebende und histologische Verfahren:
- Anamnese, Inspektion und Palpation inklusive der zervikalen Lymphknoten
- Sonographie der Halsweichteile
- Magnetresonanztomographie (MRT) als bildgebendes Verfahren der Wahl zur Beurteilung der lokalen Ausdehnung und perineuralen Ausbreitung
- Computertomographie (CT) insbesondere bei Knocheninfiltration
- Feinnadelpunktion bzw. Stanzbiopsie zur histologischen Sicherung[4]
- Staging mit CT-Thorax zum Ausschluss von Lungenmetastasen
Differentialdiagnose
Aufgrund der variablen Morphologie und des langsamen Wachstums sind verschiedene Entitäten abzugrenzen:
- pleomorphes Adenom (gutartig, jedoch ähnliche Stromaarchitektur in der Zytologie)
- basalzelliges Adenom bzw. basalzelliges Adenokarzinom (ähnliche biphasische Morphologie, fehlende Invasion)
- Mukoepidermoidkarzinom (muzinöse Komponente, MAML2-Rearrangement)
- polymorphes Adenokarzinom (insbesondere bei palatinaler Lokalisation)
Immunhistochemisch und molekularpathologisch hilft der Nachweis des MYB-NFIB-Fusionsgens zur Abgrenzung.
Therapie
Die Therapie richtet sich nach Lokalisation, Ausdehnung und Metastasierungsstatus.[5]
Operative Therapie
Therapie der Wahl ist die vollständige Resektion mit histologisch tumorfreien Resektionsrändern (R0) und intraoperativer Schnellschnittdiagnostik. Aufgrund der perineuralen Ausbreitung sind R0-Resektionen häufig schwierig zu erzielen. Bei klinischem oder radiologischem Verdacht auf Lymphknotenmetastasen erfolgt eine Neck dissection.
Strahlentherapie
Eine postoperative Strahlentherapie wird aufgrund der hohen Lokalrezidivrate und der häufigen perineuralen Infiltration in der Regel empfohlen. Als Standardverfahren gilt die intensitätsmodulierte Strahlentherapie (IMRT). Bei inoperablen Tumoren oder R1/R2-Situationen kommen in ausgewählten Fällen Protonen- oder Schwerionen-(Kohlenstoffionentherapie)Verfahren zum Einsatz. Die Strahlentherapie kann auch palliativ eingesetzt werden.
Systemische Therapie
Eine klassische Chemotherapie ist aufgrund der geringen Proliferationsrate meist wenig wirksam. In der metastasierten oder rezidivierten Situation werden Tyrosinkinaseinhibitoren wie Lenvatinib oder Axitinib eingesetzt, die jedoch eher eine Krankheitsstabilisierung als eine Tumorverkleinerung bewirken.[6] Weitere Substanzen, u.a. NOTCH-Inhibitoren bei NOTCH1-Mutationen, befinden sich in klinischer Prüfung.
Prognose
Das adenoid-zystische Karzinom zeichnet sich durch einen langsamen, aber anhaltend progredienten Verlauf mit hoher Lokalrezidivrate und später Fernmetastasen, bevorzugt in Lunge und Knochen, aus. Prognostisch ungünstige Faktoren sind:
- solides Wachstumsmuster
- perineurale Infiltration
- positive Resektionsränder
- fortgeschrittenes T-Stadium
- Fernmetastasen
Die 5-Jahres-Überlebensrate liegt insgesamt bei etwa 60 bis 70 %, die 10-Jahres-Überlebensrate jedoch deutlich niedriger (ca. 40 %), da viele Patienten späte Rezidive und Metastasen entwickeln. In der metastasierten Situation ist in der Regel nur eine palliative Therapie möglich.
Quellen
- ↑ Lorini L et al. Curative approaches for adenoid cystic carcinoma: a multifaceted disease. Oral Oncol. 2026;176:107911.
- ↑ Persson M et al. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc Natl Acad Sci USA. 2009;106(44):18740–4.
- ↑ Wagner VP et al. MYB-NFIB fusion transcript in adenoid cystic carcinoma: Current state of knowledge and future directions. Crit Rev Oncol Hematol. 2022;176:103745.
- ↑ Saoud C et al. Pitfalls in Salivary Gland Cytology. Acta Cytol. 2024;68(3):194–205.
- ↑ S3-Leitlinie Diagnostik und Therapie der Speicheldrüsenkarzinome. AWMF-Registernummer 007-120OL. Stand: aktuelle Version. https://www.awmf.org/leitlinien/detail/ll/007-120OL.html
- ↑ Hintze JM, Chintakuntlawar A. Systemic Therapy for Salivary Gland Cancers: A Review of Targeted and Chemotherapeutic Approaches. Curr Treat Options Oncol. 2026;27(1):7.