Osteogenesis imperfecta









Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
Loslegenvon altgriechisch: ὀστέον ("osteon") - Knochen und γένεσις ("genesis") - Entstehung, Ursprung
Synonyme: Glasknochenerkrankung, Morbus Lobstein
Englisch: brittle bone disease
Definition
Die Osteogenesis imperfecta, kurz OI, ist eine erbliche Erkrankung des Bindegewebes, die sich durch eine unvollständige Knochenbildung mit erhöhter Brüchigkeit der Knochen auszeichnet.
Epidemiologie
Die Häufigkeit des Auftretens beträgt 1: 10.000 – 15.000, in Deutschland sind ca. 4.000 – 5.000 Menschen betroffen.
Ätiologie
Ursache der Osteogenesis imperfecta sind verschiedene Störungen der Biosynthese von Kollagen, einem sehr wichtigen Bestandteil der Knochenmatrix. In 95 % der Fälle sind Gene betroffen, die zur Synthese des Kollagens Typ I wichtig sind (COL1A1 und COL1A2). Kommt es zu einem Verlust eines COL1A1-Allels, so ist nur eine verminderte Synthese von Kollagen Typ I möglich. Das Kollagen ist jedoch intakt, so dass die Ausprägung der Erkrankung mild ist.
Kommt es jedoch zu Mutationen im COL1A1- oder COL1A2-Gen (z.B. durch Punktmutation, oder alternatives Splicing), so wird überwiegend defektes und nur wenig intaktes Kollagen vom Typ I hergestellt. Meist geschieht dies durch Substitution der wichtigsten Aminosäure Glycin in der Tripelhelix des Kollagens durch eine andere Aminosäure. Zudem ist oft die Verdrillung der Kollagen-Tripelhelix gestört, was zu einer verminderten Stabilität führt.
Durch diesen dominant-negativen Effekt kommt es zu einer schweren Ausprägung (Phänotyp) der Erkrankung.
Einteilung
Die Einteilung verschiedener Formen der Osteogenesis imperfecta ist umstritten. Es existieren mehrere Einteilungssysteme, die unterschiedliche Kriterien anlegen.
... nach klinisch-radiologischen Kriterien
Nach der erweiterten Sillence-Klassifikation (2014) unterscheidet man folgende Typen der Osteogenesis imperfecta:[1]
| Typ | Klinik | betroffene Gene | Erbgang |
|---|---|---|---|
| I* | leichter Verlauf ohne Deformierungen, blaue Skleren | COL1A1, COL1A2 | AD |
| II** | Sehr schwerer, meist perinatal letaler Verlauf (vielfache Knochenbrüche mit kongenitaler Deformierung, Minderwuchs), blaue Skleren | COL1A1, COL1A2, seltener andere (CRTAP, LEPRE1, PPIB) | AD, AR |
| III | mittelschwerer, progressiv-deformierender Verlauf, blaue Skleren | COL1A1, COL1A2, andere (z.B. BMP1, CRTAP, LEPRE1, SERPINH1) | AR, seltener AD |
| IV | variabler Verlauf ohne blaue Skleren | COL1A1, COL1A2, andere (z.B. WNT1, CRTAP, PPIB, SP7) | AD, seltener AR oder XR |
| V | moderater bis schwerer Verlauf mit Ossifikation der Membrana interossea cruris und antebrachii, Neigung zu hyperplastischem Kallus | IFITM5 | AD |
* früher: Hoeve-Syndrom, Lobstein-Krankheit, ** früher: Vrolik-Krankheit
Aufgrund der Uneinheitlichkeit von ursächlichem Gen und Erbgang innerhalb der Sillence-Typen ist das Klassifikationssystem umstritten.
... nach Pathomechanismus
Die Klassifikation von Forlino et al. unterscheidet nach zugrundeliegendem Pathomechanismus vier Typen der Osteogenesis imperfecta:[2][3]
- Typ A: Defekte von Kollagensynthese, -struktur oder -prozessierung
- Typ B: Defekte der posttranslationalen Kollagenmodifikation
- Typ C: Defekte der Knochenmineralisation
- Typ D: Defekte der Osteoblastenentwicklung
... nach genetischer Grundlage
Die OMIM-Datenbank unterscheidet nach zugrundeliegender Genmutation insgesamt 22 Formen der Osteogenesis imperfecta (I-XXII). Die klinisch-radiologischen Typen I-V aus der erweiterten Sillence-Klassifikation wurden hierbei übernommen, jedoch auf COL1A1/2- und IFITM5-Gendefekte beschränkt.[3]
Symptome
Viele Symptome der Osteogenesis imperfecta betreffen das Skelettsystem:
- Multiple Frakturen (Knochenbrüche)
- Deformierung der Schädelkalotte
- Ausbildung von flachen Wirbelkörpern (Platyspondylie)
- Verzögerung der Ossifikation
- Kleinwuchs
- Verformung der Wirbelsäule (Skoliose, Kyphose)
Darüber hinaus gibt es Symptome, die nicht das Skelett, sondern andere Organsysteme betreffen, zum Beispiel:
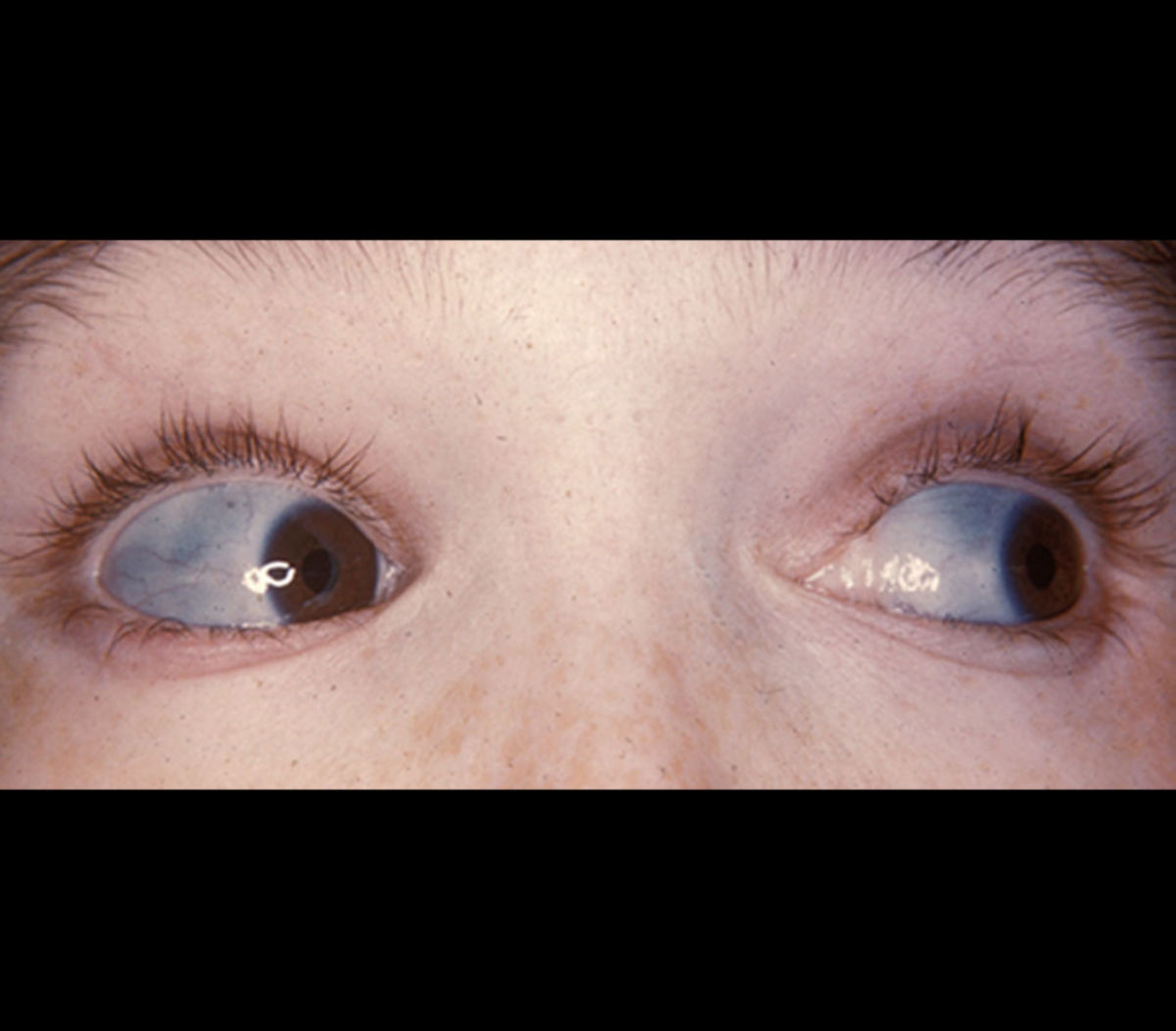
- Blaue Skleren
- Schallleitungsschwerhörigkeit
- Dentinogenesis imperfecta (Störung der Zahnbildung)
- Überstreckbarkeit der Gelenke
- Neigung zu Hämatomen
- Aortendilatation
- Aorteninsuffizienz
- Subdurales Hämatom
- schwache Muskulatur
- übermäßiges Schwitzen
- Neigung zu Leistenbrüchen
- Myopie (Kurzsichtigkeit)
Im Röntgenbild zeigt sich eine erhöhte Transparenz des Knochens, da zu wenig schattengebende Knochensubstanz vorhanden ist.
Therapie
Die genetischen Ursachen der Osteogenesis imperfecta können derzeit (2024) nicht behandelt werden, die Therapie ist symptomatisch ausgerichtet.
Es wird versucht, den Krankheitsverlauf mit Calcitonin, Calciferol, Fluor und Bisphosphonaten zu verlangsamen und abzumildern. Die Frakturen werden chirurgisch und orthopädisch versorgt. Durch Marknagelung lässt sich die Stabilität der Knochen erhöhen. Zudem ist eine Physiotherapie wichtig, um weiterem Knochenabbau vorzubeugen und durch Muskelaufbau die Sturzgefahr zu vermindern.
Quiz
Bildquelle
- Bildquelle für Quiz: ©Cara Shelton / Unsplash
Einzelnachweise
- ↑ Van Dijk, Sillence. Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment. American Journal of Medical Genetics, Part A, 2014.
- ↑ Forlino A, Marini JC. Osteogenesis imperfecta. Lancet, 2016.
- ↑ 3,0 3,1 Panzaru et al. Classification of osteogenesis imperfecta: Importance for prophylaxis and genetic counseling. World Journal of Clinical Cases, 2023.