Apert-Syndrom: Unterschied zwischen den Versionen
Keine Bearbeitungszusammenfassung |
Keine Bearbeitungszusammenfassung |
||
| Zeile 17: | Zeile 17: | ||
==Ätiologie== | ==Ätiologie== | ||
Die Ursache des Apert-Syndroms ist eine [[Mutation]] im [[FGFR2]]-[[Gen]] | Die Ursache des Apert-Syndroms ist eine [[Mutation]] im [[FGFR2]]-[[Gen]] auf dem [[Chromosom 10]], Genlocus: 10q26, das für den [[Fibroblasten-Wachstumsfaktor-Rezeptor 2]] codiert. Die Vererbung erfolgt [[autosomal-dominant]]. Es besteht eine vollständige [[Penetranz]], das heißt, jeder Träger des mutierten Gens ist auch von der Erkrankung betroffen. Die [[Varianz]] des Apert-Syndroms ist sehr hoch, das bedeutet, die Symptome sind bei den Betroffenen unterschiedlich stark ausgeprägt. | ||
Die Vererbung erfolgt [[autosomal-dominant]]. Es besteht eine vollständige [[Penetranz]], das heißt, jeder Träger des mutierten Gens ist auch von der Erkrankung betroffen. Die [[Varianz]] des Apert-Syndroms ist sehr hoch, das bedeutet, die Symptome sind bei den Betroffenen unterschiedlich stark ausgeprägt. | |||
Häufig sind Neumutationen Ursache des Apert-Syndroms. Hierbei scheint das Alter des Vaters eine Rolle zu spielen. Das Apert-Syndrom tritt häufiger bei Kindern älterer Väter auf. Etwa eines von 130.000 Neugeborenen ist von der Erkrankung betroffen. | Häufig sind Neumutationen Ursache des Apert-Syndroms. Hierbei scheint das Alter des Vaters eine Rolle zu spielen. Das Apert-Syndrom tritt häufiger bei Kindern älterer Väter auf. Etwa eines von 130.000 Neugeborenen ist von der Erkrankung betroffen. | ||
Version vom 10. Juni 2016, 15:41 Uhr
nach Eugène Apert, Pädiater aus Paris (1868-1940)
Synonym: Akrozephalosyndaktylie-Syndrom
Definition
Das Apert-Syndrom ist eine Erbkrankheit, die zu den Akrozephalosyndaktylie-Syndromen gehört und zu multiplen Fehlbildungen führt.
ICD10-Code: Q87.0
Hintergrund
Das Apert-Syndrom wird zu den kraniofazialen Fehlbildungen gerechnet. Zu dieser Gruppe gehören außerdem folgende Erkrankungen:
Das Apert-Syndrom ist die schwerwiegendste Erkrankung dieser Gruppe.
Ätiologie
Die Ursache des Apert-Syndroms ist eine Mutation im FGFR2-Gen auf dem Chromosom 10, Genlocus: 10q26, das für den Fibroblasten-Wachstumsfaktor-Rezeptor 2 codiert. Die Vererbung erfolgt autosomal-dominant. Es besteht eine vollständige Penetranz, das heißt, jeder Träger des mutierten Gens ist auch von der Erkrankung betroffen. Die Varianz des Apert-Syndroms ist sehr hoch, das bedeutet, die Symptome sind bei den Betroffenen unterschiedlich stark ausgeprägt.
Häufig sind Neumutationen Ursache des Apert-Syndroms. Hierbei scheint das Alter des Vaters eine Rolle zu spielen. Das Apert-Syndrom tritt häufiger bei Kindern älterer Väter auf. Etwa eines von 130.000 Neugeborenen ist von der Erkrankung betroffen.
Klinik
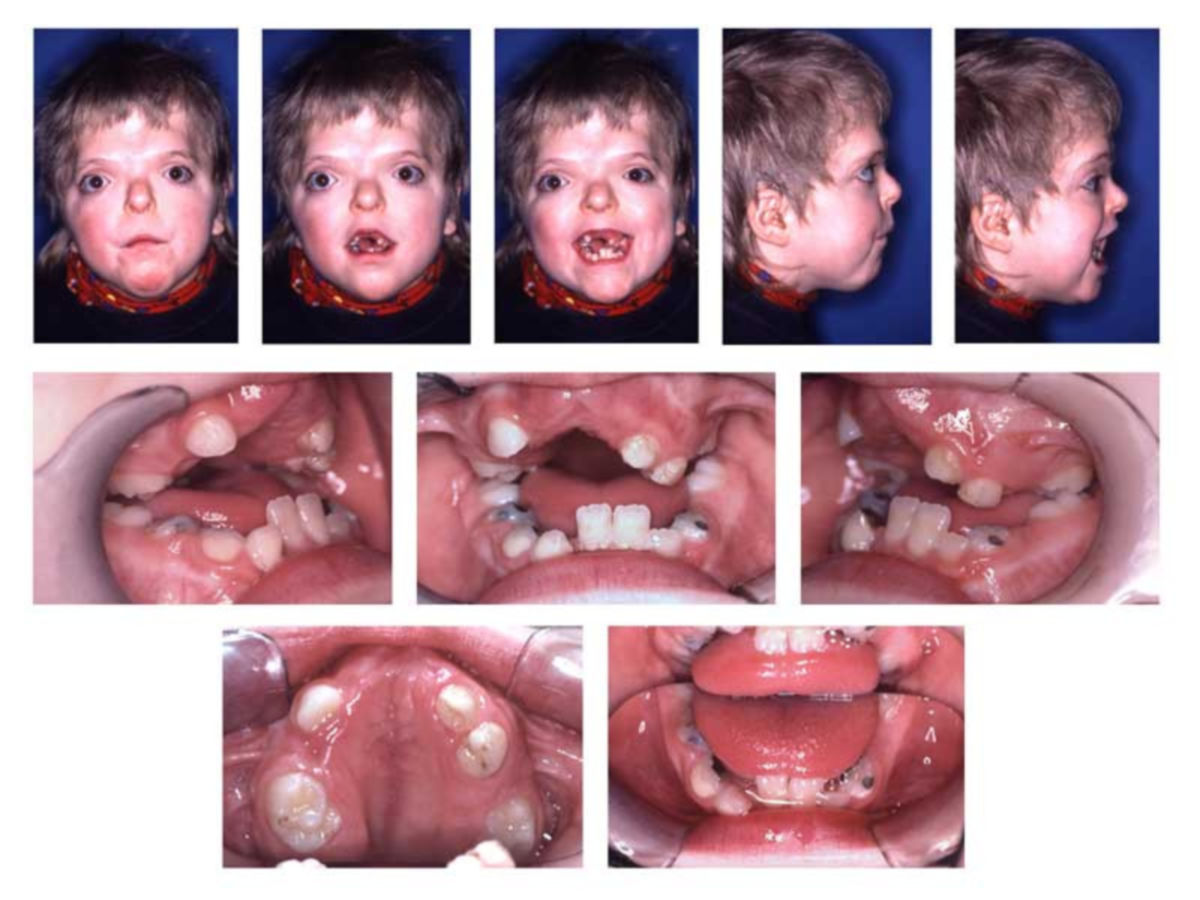
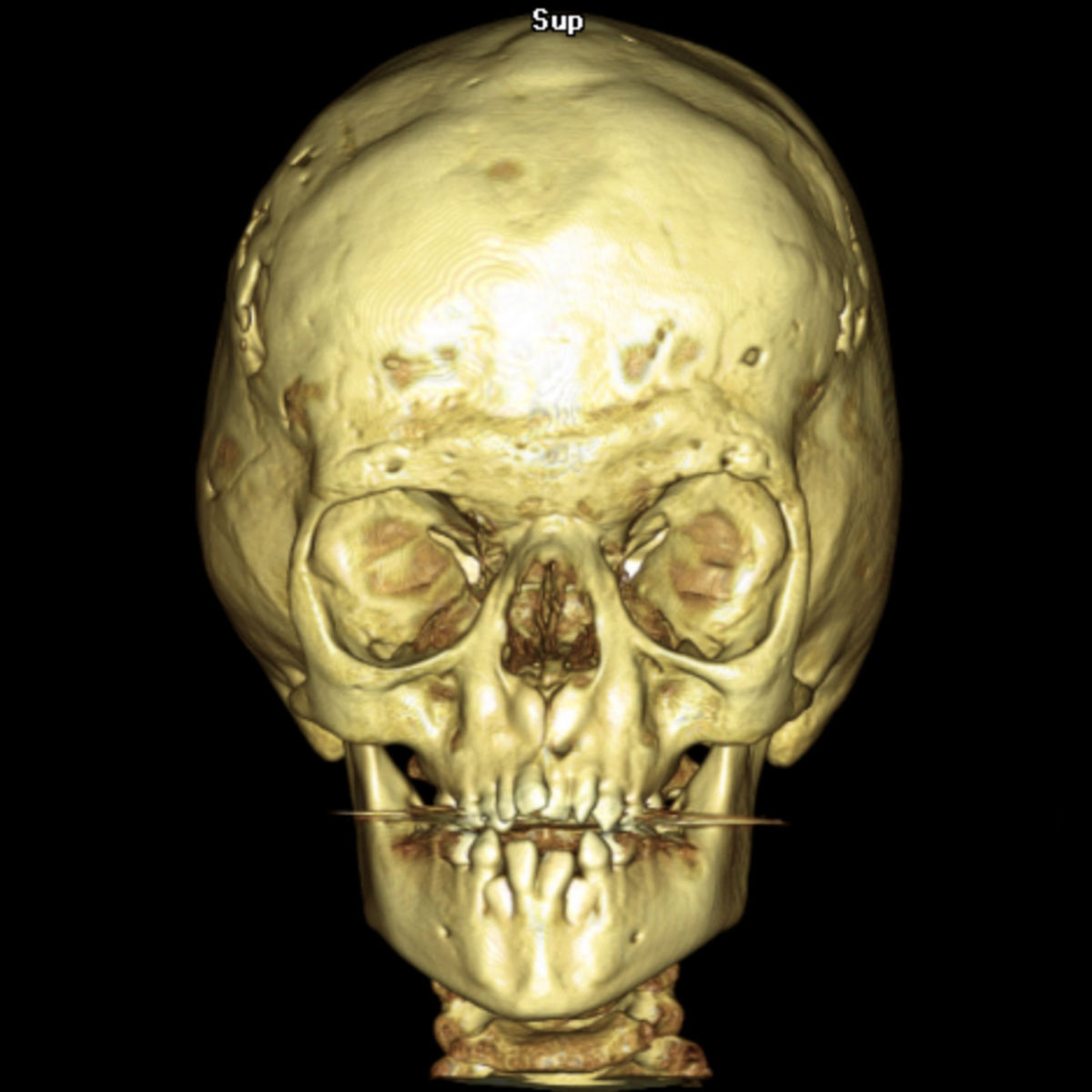
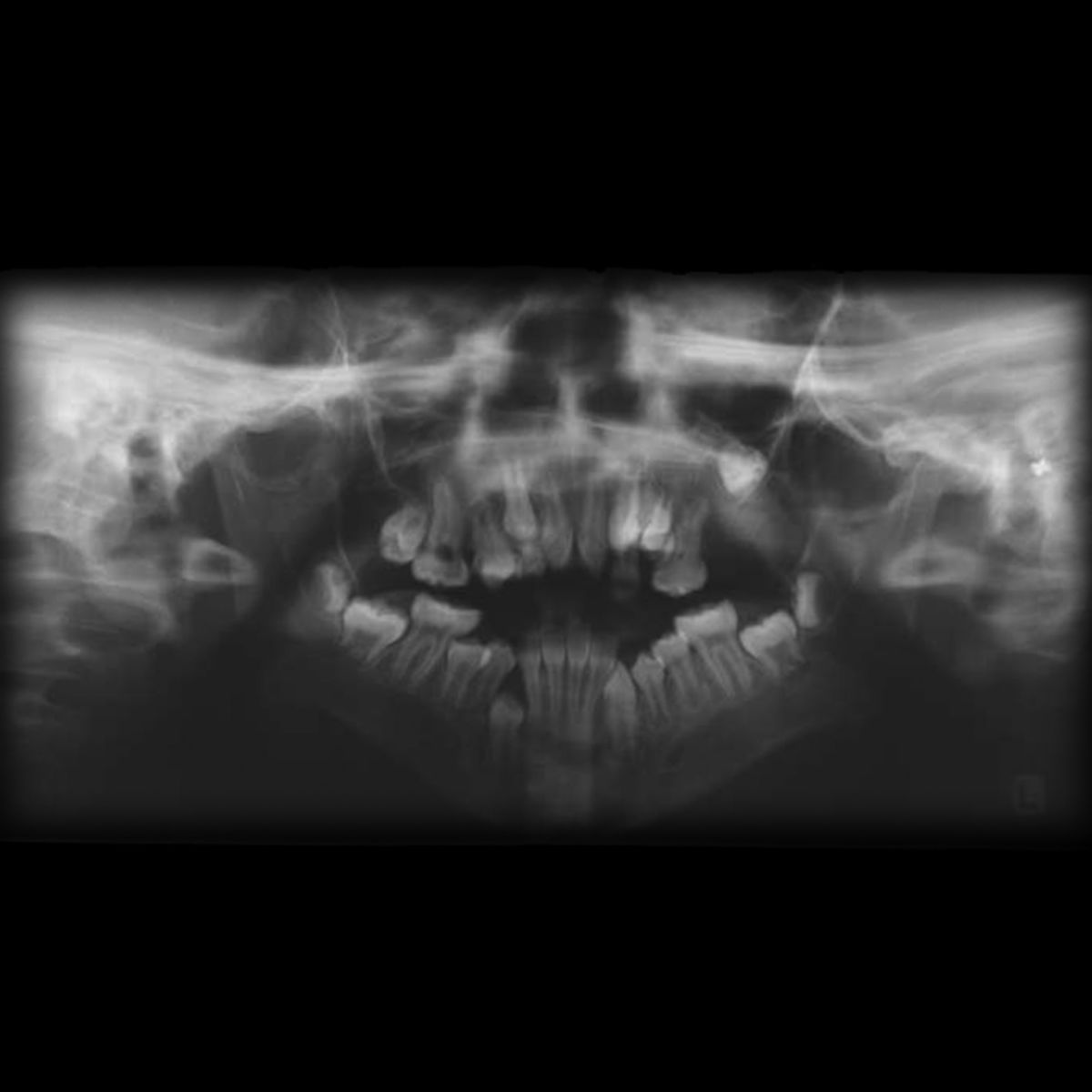
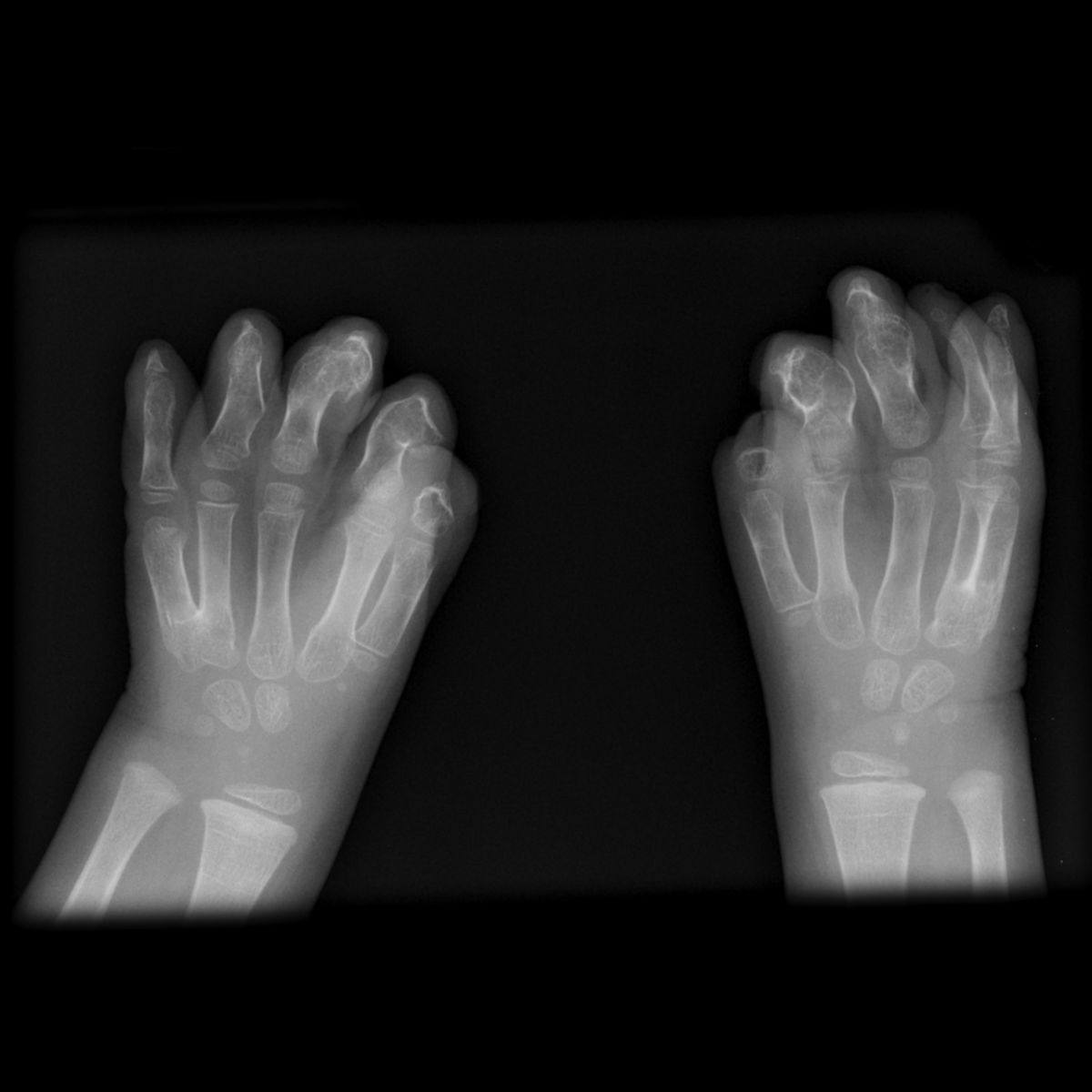
Es gibt mehrere Symptome, die häufig im Rahmen des Apert-Syndroms auftreten:
- Fehlbildung des Kopfes
- Turmschädel durch vorzeitige Verknöcherung der Koronarnähte des Schädels
- Fehlbildungen des Gesichts
- flache Orbita
- Exophthalmus: Hervorstehender Augapfel
- Hypertelorismus: Vergrößerter Augenabstand
- Fehlbildungen des Oberkiefers
- Gaumenspalte
- Fehlbildungen der Hände und Füße
- Syndaktylie: Finger (und Zehen) sind nicht getrennt, sondern miteinander verwachsen. Diese Fehlbildung tritt beim Apert-Syndrom immer symmetrisch auf.
- Versteifung der Mittelgelenke der Finger (z.T. fehlen diese vollständig)
- Löffelhände
- Synostosen
- Symphalangie
- Brachydaktylie
- oft gemeinsame Fingernägel
- Skoliose: Wirbelsäulenverkrümmung
- Schwerhörigkeit
- Fehlsichtigkeit
- Probleme bei der Atmung
- Geistige Retardierung (in ca. 80% der Fälle)
- Hydrozephalus: Dieser entsteht bei zu hohem Hirndruck infolge der verwachsenen Schädelknochen
Diagnostik
Das Apert-Syndrom kann schon vor der Geburt im Rahmen der Pränataldiagnostik auffallen. Häufig sind typische Merkmale der Erkrankung im Feinultraschall zu erkennen (ab ca. 3. Schwangerschaftsmonat). Die Diagnose erfolgt dann meist durch Fruchtwasseruntersuchung, die seit neuerer Zeit schon ab der 8. Schwangerschaftswoche möglich ist. Nach der Geburt geben die typischen Symptome Hinweis auf die Erkrankung, die Bestätigung der Diagnose ist durch molekulargenetische Methoden möglich.
Therapie
Zur Behandlung der Schädel-Deformität ist oftmals eine chirurgische Sprengung der Schädelnähte notwendig. Außerdem wird die Syndaktylie chirurgisch getrennt und danach eine Stellungskorrektur der Finger und Zehen vorgenommen. Manchmal muss auch das Mittelohr operiert werden, weil Gehörknöchelchen beim Apert-Syndrom z.T. nicht ausgebildet sind.
Zudem darf die psychische Belastung der betroffenen Kinder nicht vernachlässigt werden: Das "Anders-sein" führt oftmals zur Ablehnung durch die Umwelt. Dazu kommen die vielen Operationen, die die Kinder schon in den ersten Lebensjahren mitmachen müssen. All dies führt bei den Betroffenen oft zu psychischen Problemen, deren Behandlung auch Teil der Therapie sein sollte.