ANCA-assoziierte Vaskulitis







Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenSynonym: ANCA-assoziierte Kleingefäßvaskulitis
Englisch: ANCA-associated vasculitis
Definition
Die ANCA-assoziierten Vaskulitiden, kurz AAV, umfassen eine Gruppe von nekrotisierenden Gefäßentzündungen mit prädominantem Befall kleiner Gefäße, wie z.B. der kleinen Arterien, Arteriolen, Venolen und Kapillaren. Diese Erkrankungen sind häufig – aber nicht immer – mit antineutrophilen zytoplasmatischen Antikörpern (ANCA) assoziiert.
Einteilung
Die ANCA-assoziierten Vaskulitiden umfassen gemäß der Chapel-Hill-Nomenklatur von 2012 die folgenden Krankheiten:[1]
Epidemiologie
Die AAV zählen zu den seltenen Erkrankungen. Die Inzidenz beträgt je nach Entität ca. 2–10 Fälle pro 1.000.000 Einwohner pro Jahr, wobei die GPA in Nordeuropa am häufigsten vertreten ist, während die MPA in Südeuropa und Asien überwiegt.[2] Der Erkrankungsgipfel liegt im 6.–7. Lebensjahrzehnt. Männer und Frauen sind etwa gleich häufig betroffen.
Pathophysiologie
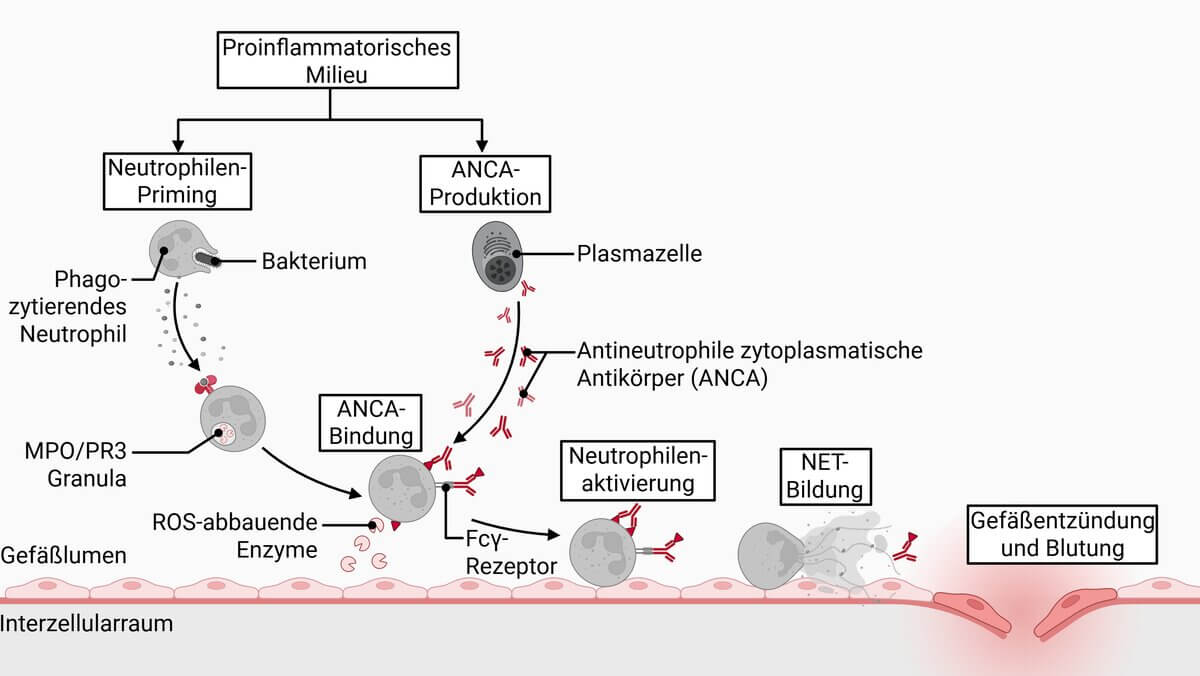
Pathogenetisch zentral ist die Autoantikörper-vermittelte Aktivierung von Neutrophilen. Die ANCA richten sich gegen zwei Hauptantigene:[3]
- PR3-ANCA (Proteinase 3): typischerweise assoziiert mit GPA, c-ANCA-Muster in der Immunfluoreszenz
- MPO-ANCA (Myeloperoxidase): typischerweise assoziiert mit MPA und EGPA, p-ANCA-Muster in der Immunfluoreszenz
Die Bindung der ANCA an vorzeitig aktivierte Neutrophile führt zu deren vollständiger Aktivierung mit konsekutiver Ausschüttung von proteolytischen Enzymen und reaktiven Sauerstoffspezies. Dies bewirkt eine nekrotisierende Gefäßwandentzündung. Zusätzlich trägt die Aktivierung des Komplementsystems (Lektin- und Alternativweg) zur Gewebeschädigung bei. Bei GPA und EGPA spielen außerdem granulomatöse Entzündungsreaktionen eine pathogenetische Rolle.
Klinik
Wegweisende Befunde sind:
- pulmorenales Syndrom
- Glomerulonephritis
- chronische destruktive Erkrankung des oberen Respirationstrakts
- chronische Sinusitis, chronische Otitis
- pulmonale Rundherde und granulomatöse interstitielle Lungenerkrankung
- diffuse alveoläre Hämorrhagie mit Kapillaritis
- interstitielle Lungenerkrankung mit UIP-Muster (bei MPO-ANCA)
- Hautvaskulitis mit Zeichen einer Systemerkrankung
- retroorbitales Granulom
- Skleritis, Episkleritis
- Mononeuritis multiplex, periphere Neuropathie
Diagnostik
Die Diagnose stützt sich auf eine Kombination aus Klinik, Serologie, Bildgebung und – sofern möglich – Biopsie.[3]
Serologie
- ANCA-Bestimmung mittels indirekter Immunfluoreszenz (IIF) und Antigen-spezifischem ELISA (PR3-ANCA, MPO-ANCA)
- Blutbild, BSG, CRP, Kreatinin, Urinstatus (Hämaturie, Proteinurie)
- Eosinophilie bei EGPA (typischerweise >10 % oder > 1.500/µl)
Cave: Ein negativer ANCA-Befund schließt eine AAV nicht aus – ca. 10 % der Fälle verlaufen ANCA-negativ.
Bildgebung
- Computertomographie des Thorax: pulmonale Rundherde, Kavernen, Milchglasinfiltrate, interstitielle Veränderungen
- Computertomographie der Nasennebenhöhlen: chronisch-destruktive Veränderungen bei GPA
Biopsie
Die Sicherung der Diagnose erfolgt durch den Nachweis einer nekrotisierenden Vaskulitis mit oder ohne Granulombildung, je nach biopsierten Organ.
Therapie
Die Therapie der AAV gliedert sich in Remissionsinduktion und Remissionserhaltung. Grundlage sind die S3-Leitlinie der DGRh sowie die EULAR-Empfehlungen 2022.[4]
Remissionsinduktion
Bei organgefährdender oder lebensbedrohlicher Erkrankung (GPA, MPA):
- Rituximab in Kombination mit hochdosierten Glukokortikoiden – Mittel der Wahl
- Cyclophosphamid als Alternative, insbesondere bei eingeschränkter Nierenfunktion oder fehlendem Ansprechen auf Rituximab
Bei EGPA mit nicht organgefährdender Erkrankung genügen oft Glukokortikoide allein. Bei rezidivierendem oder refraktärem Verlauf sind IL-5-Inhibitoren (z.B. Mepolizumab) zugelassen.
Remissionserhaltung
Nach erfolgreicher Remissionsinduktion:
- Rituximab (bevorzugt) oder Azathioprin für mindestens 24 Monate
- Niedrigdosis-Glukokortikoide mit dem Ziel der schrittweisen Dosisreduktion
- Bei EGPA: Mepolizumab zur steroidreduzierenden Erhaltungstherapie
Prognose
Unbehandelt verlaufen GPA und MPA rasch progredient mit hoher Mortalität. Unter moderner Immunsuppression erreichen > 80 % der Patienten eine Remission. Das 5-Jahres-Überleben liegt bei über 75 %. Das Rezidivrisiko ist bei PR3-ANCA-positiven Patienten (GPA) höher als bei MPO-ANCA-positiven Patienten (MPA). Häufige Langzeitkomplikationen umfassen chronische Niereninsuffizienz, therapiebedingte Infektionen und Malignome.
Quellen
- ↑ Jennette et al., 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides, Arthritis Rheum, 2013
- ↑ Yaseen und Mandell, ANCA associated vasculitis (AAV): a review for internists, Postgrad Med, 2022
- ↑ 3,0 3,1 Schreiber und Reimers, Vaskulitiden und Anti-GBM-Erkrankung, Innere Medizin (Heidelb), 2026
- ↑ AWMF, S3-Leitlinie Diagnostik und Therapie der ANCA-assoziierten Vaskulitiden, AWMF-Registernummer 060-012, Version 1.1. 2024