Akromegalie










Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenSynonym: Hyperpituitarismus
Englisch: acromegaly
Definition
Akromegalie ist eine endokrinologische Erkrankung, die durch eine Überproduktion des Wachstumshormons Somatotropin (STH) im Hypophysenvorderlappen (HVL) gekennzeichnet ist.
Epidemiologie
Bei der Akromegalie handelt es sich insgesamt um eine eher seltene Erkrankung mit einer Inzidenz von etwa 3–5 Fällen pro 1 Million Einwohner pro Jahr. Am häufigsten tritt die Erkrankung zwischen dem 45. und 50. Lebensjahr auf.
Physiologie
Die Sekretion von Somatotropin aus dem Hypophysenvorderlappen wird vom Hypothalamus durch zwei Einflussgrößen gesteuert:
- Das im Hypothalamus gebildete GHRH wird über das hypothalamo-hypophysäre Portalsystem zum HVL transportiert und stimuliert dort die Synthese und Freisetzung von Somatotropin.
- Verschiedene Körpergewebe, darunter das endokrine Pankreas produzieren als Antwort auf die Somatotropin-Ausschüttung das Hormon Somatostatin, das in Form einer negativen Rückkoppelung die Ausschüttung von GHRH hemmt.
Somatotropin beeinflusst das Wachstum nicht direkt, sondern stimuliert wiederum die Produktion von IGF-1 (insulinlike growth factor-I) in der Leber, das dann die Wachstumseffekte in den Geweben auslöst.
Ursachen
Symptome
Die Symptomatik der Akromegalie wird entscheidend davon geprägt, ob die Erkrankung vor oder nach dem Verschluss der Epiphysenfugen eintritt:
Vor Abschluss des Längenwachstums
Vor Abschluss des Längenwachstums kommt es zum sog. Gigantismus bzw. hypophysären Riesenwuchs. Die normalen Körperproportionen bleiben dabei weitgehend erhalten.
Nach Abschluss des Längenwachstums
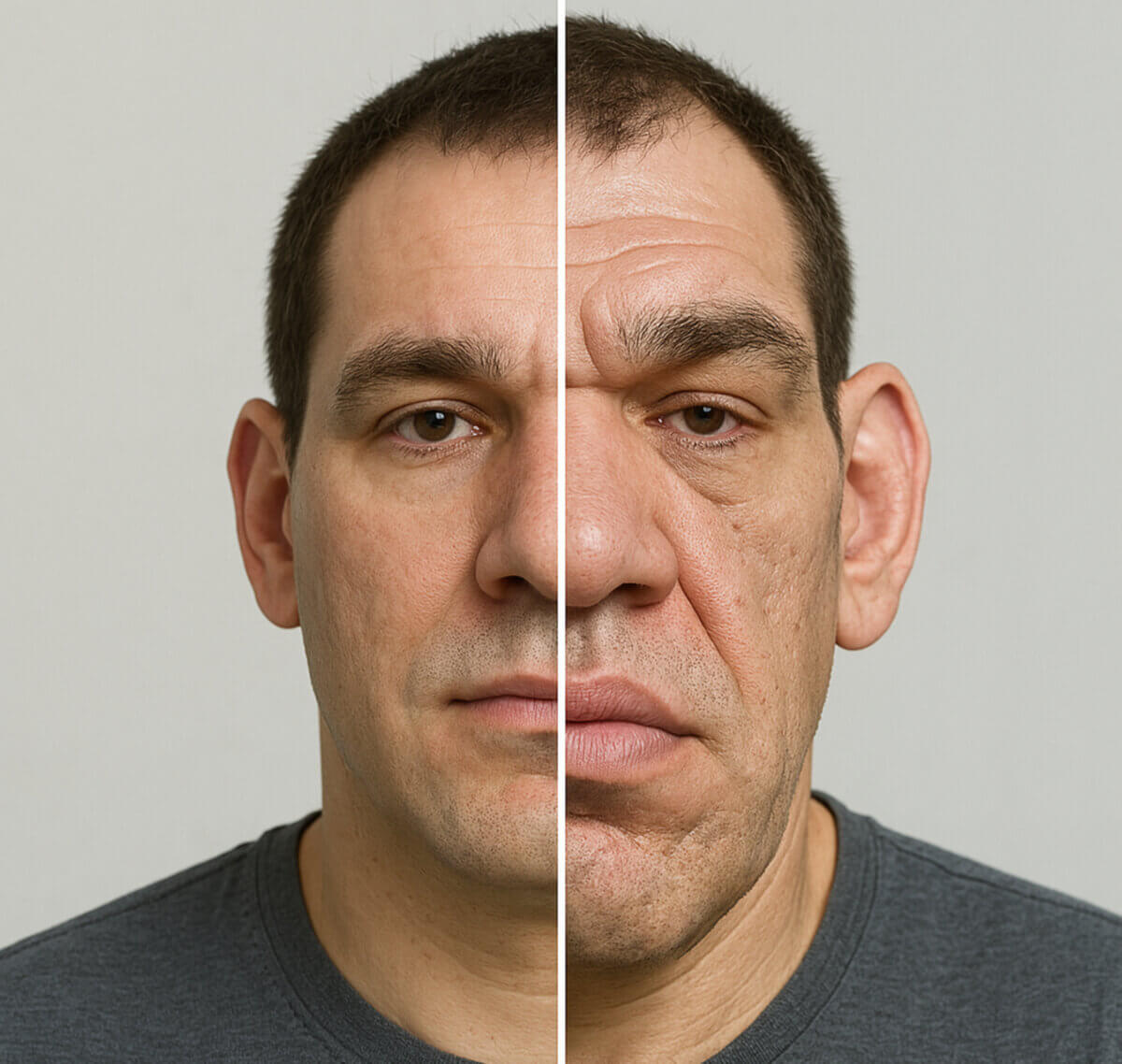
Nach Verschluss der Epiphysenfugen ist ein Wachstum nur noch an den knöchernen Akren (Nase, Finger, Zehen, Kinn und Jochbogen) und im Bereich der Weichteile möglich. Die Körperproportionen wirken durch dieses Wachstum unharmonisch und deutlich vergröbert. Dem Patienten fällt auf, dass Kleidungsstücke (z.B. Schuhe und Ringe) zu klein werden. Weitere, typische Symptome sind:
- Makroglossie
- Kopfschmerzen
- Hypertonie
- Hyperhidrose
- Hypertrichose
- Arthropathien
- Hypästhesien, Parästhesien
- Sehstörungen, Gesichtsfeldeinschränkungen (bei suprasellärer Ausbreitung des Adenoms)
- Okklusionsstörungen
Die Überproduktion der Wachstumshormone löst bei rund 1/4 der Patienten einen Diabetes mellitus aus. Ursächlich sind Störungen des Glukosestoffwechsels.
Auch die inneren Organe des Patienten können vergrößert sein, z.B. die Abdominalorgane (Viszeromegalie) oder das Herz (Kardiomegalie). Die Kardiomegalie ist dabei meistens eher die Folge einer infolge des STH-Überschuss auftretenden Hypertonie.
Durch die Makroglossie kann es zu einem sekundären Schlafapnoe-Syndrom kommen. Insgesamt kommt es zu einer Verdickung der Schleimhäute des Nasen-Rachen-Raums, was die Atmung erschwert.
Bedingt durch eine Pachydermie (Verdickung von Haut und Bindegewebe) kommt es zur Vergröberung der Gesichtszüge und zu Veränderungen an Händen und Füßen.
Wird die Akromegalie durch einen Hypophysen-Tumor verursacht, so können Symptome des sekundären Hypogonadismus (sekundäre Amenorrhö und Zyklusstörungen bei Frauen; Libido- und Potenzverlust bei Männern) auftreten. Sezerniert der Tumor zusätzlich Prolaktin (ca. 1/5 der Fälle), tritt eine Hyperprolaktinämie auf.
Patienten mit STH-Überschuss haben außerdem ein erhöhtes Risiko, an Kolontumoren zu erkranken.
Diagnostik
Die Akromegalie verläuft schleichend, was die Diagnosestellung erschwert und häufig zu einer Fehlinterpretation der Symptome führt. Neben dem typischen klinischen Bild im Spätstadium der Erkrankung ist in den frühen Stadien vor allem die Hormonanalyse richtungsweisend:
- Somatotropin (Wachstumshormon, STH) im Serum
- IGF-1
- STH-Suppressionstest (Serum-STH nach Glukosebelastung)
Dabei muss die zirkardiane Rhythmik der Hormone im Blut berücksichtigt werden.
Zur Lokalisationsdiagnostik (Tumornachweis) dienen zusätzlich CT und MRT.
Therapie
Chirurgisch
Die Therapie besteht in der chirurgischen Entfernung des Tumors der Hirnanhangsdrüse. Für die Operation stehen ein transsphenoidaler (durch die Nase und Teile der Nasennebenhöhlen) und ein transkranieller Zugang (durch die Schädeldecke) zur Verfügung.
Medikamentös
Nach einer Operation wird eine medikamentöse Behandlung erforderlich, wenn das hormonproduzierende Gewebe nicht vollständig entfernt werden konnte. Mehrere Wirkstoffgruppen stehen für die medikamentöse Behandlung zur Verfügung: der Dopaminagonist Cabergolin (Cabaseril®), die Somatostatinanaloga Octreotid (Sandostatin®) und Lanreotid (Somatoline Autogel®) sowie der Wachstumshormonantagonist Pegvisomant (Somavert®). Gegebenenfalls kann bei einem großen Tumor eine medikamentöse Therapie vor einer chirurgischen Entfernung angewendet werden. Das Ziel einer Vorbehandlung mit den genannten Medikamenten ist, den Tumor zu verkleinern und den Allgemeinzustand der Patienten zu verbessern.
Strahlentherapie
Eine weitere Therapieoption stellt die Strahlentherapie dar. Indikationen sind Inoperabilität oder eine unvollständige Resektion bei der Operation. Die Wirkung der Strahlentherapie kann jedoch unter Umständen erst nach Monaten oder Jahren eintreten. Bei zunehmender Dauer der Bestrahlung besteht die Gefahr einer Hypophysenvorderlappeninsuffizienz.
Bei Tumoren mit einer Größe unter 5 cm und einem Abstand von mindestens 5 mm zum Chiasma opticum besteht die Möglichkeit einer hoch dosierten Strahlentherapie. Diese sollte allerdings erst nach genauer Lokalisation des Tumors erfolgen.
Quiz
Bildquelle
- Bildquelle für Flexikon-Quiz: KI-generiert