ALA-Dehydratasemangel-Porphyrie







Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenSynonyme: Doss-Porphyrie, ALA-Defizienz-Porphyrie, ALAD-Porphyrie, Porphyrie durch ALAD-Mangel, Porphyrie durch δ-Aminolävulinsäure-Dehydratase-Mangel, ALA-Dehydratase-Defizienz-Porphyrie
Englisch: porphyria due to ALA dehydratase deficiency, porphyria of Doss
Definition
Die ALA-Dehydratasemangel-Porphyrie, kurz ALAD-Porphyrie, ist eine sehr seltene Form der akuten hepatischen Porphyrie. Die Erkrankung nimmt einen chronischen Verlauf, der von akuten Anfällen unterbrochen wird.
Epidemiologie
Die ALAD-Porphyrie gehört zu den seltenen Erkrankungen. Weltweit sind bislang (2023) nur wenige Fälle in der Literatur beschrieben. Bei diesen Fällen handelte es sich ausschließlich um männliche Patienten, bei denen sich die Erkrankung meist bis zur Adoleszenz manifestiert.
Ätiologie
Bei der ALAD-Porphyrie liegt ein autosomal-rezessiv vererbter Defekt der δ-ALA-Dehydratase (ALAD) vor. Dieses Enzym steht am Anfang der Hämsynthese und katalysiert den Syntheseschritt von zwei δ-Aminolävulinsäure-Molekülen (5-ALA) zu Porphobilinogen. Bei homozygotem Genotyp besteht meist eine Restaktivität der ALAD von weniger als 10%.
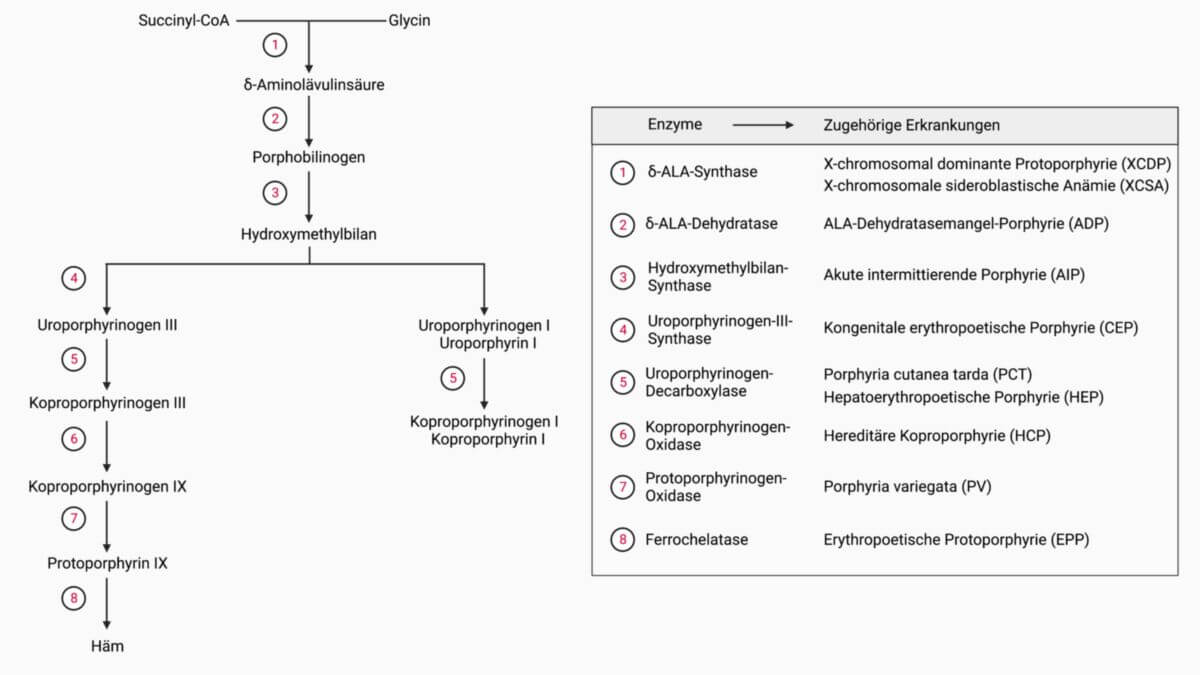
Pathophysiologie
Durch den ALAD-Defekt akkumuliert 5-ALA in den Erythrozyten und Hepatozyten und entfaltet eine toxische Wirkung.
Symptome
Das klinische Bild kann je nach Restaktivität sehr unterschiedlich sein.
Mögliche Symptome sind abdominelle Schmerzen sowie eine periphere Neuropathie und neuropsychiatrische Symptome (z.B. Desorientierung, Angst, Unruhe, Halluzinationen). Auch eine Wachstumsstörung ist möglich.
Diagnose
Die Patienten weisen neben den typischen Symptomen in akuten Phasen pink- oder rotgefärbten Urin auf. Labormedizinisch sind erhöhte 5-ALA-Level im Blut und Urin nachweisbar. Die abschließende Diagnose basiert auf der Identifikation der auslösenden Genmutation mittels molekulargenetischer Untersuchung. Es sind verschiedene Mutationen im ALAD-Gen beschrieben. Betroffene erben dann verschiedene Mutationen von jedem Elternteil (Compound-Heterozygotie).
Therapie
Derzeit (2023) existiert keine kausale Therapie der ALAD-Porphyrie, die Behandlung erfolgt rein symptomatisch. Bei akuten schweren Schüben wird Hämin eingesetzt, um die Hämsynthese temporär zu hemmen.
Quellen
- orpha.net - Porphyria due to ALA dehydratase deficiency, abgerufen am 25.05.2023
- Mohan und Madan Ala Dehydratase Deficiency Porphyria StatPearls 2022
- Suttorp et al., Harrisons Innere Medizin, Thieme Verlag, 20. Auflage, 2022
- European Porphyria Network – The porphyrias, abgerufen am 25.05.2023