Liposarkom







Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenEnglisch: liposarcoma
Definition
Das Liposarkom ist eine maligne Neoplasie des Fettgewebes.
Epidemiologie
Liposarkome machen etwa 15–20 % aller Weichteilsarkome aus und treten vor allem im höheren Erwachsenenalter auf, mit einem Häufigkeitsgipfel jenseits des 50. Lebensjahres. Männer sind etwas häufiger betroffen als Frauen.
Ätiologie
Die Entstehung von Liposarkomen erfolgt in der Regel nicht auf der Grundlage von Lipomen. Die genaue Ätiologie ist bislang (2026) unklar. Charakteristisch sind jedoch subtypspezifische genetische Veränderungen. Beispiele sind MDM2- und CDK4-Amplifikationen bei hoch differenzierten und dedifferenzierten Liposarkomen sowie FUS-DDIT3-Translokationen beim myxoiden Liposarkom.
Lokalisation
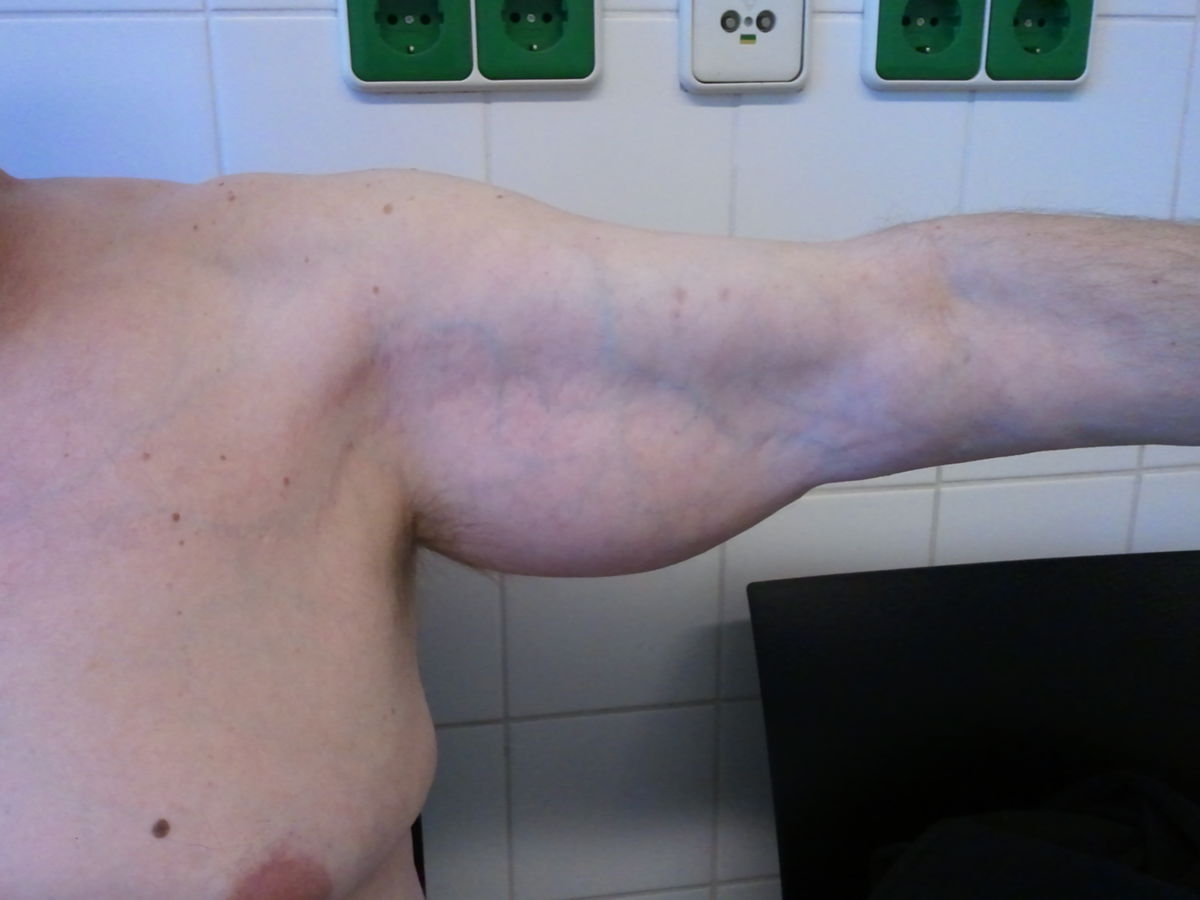
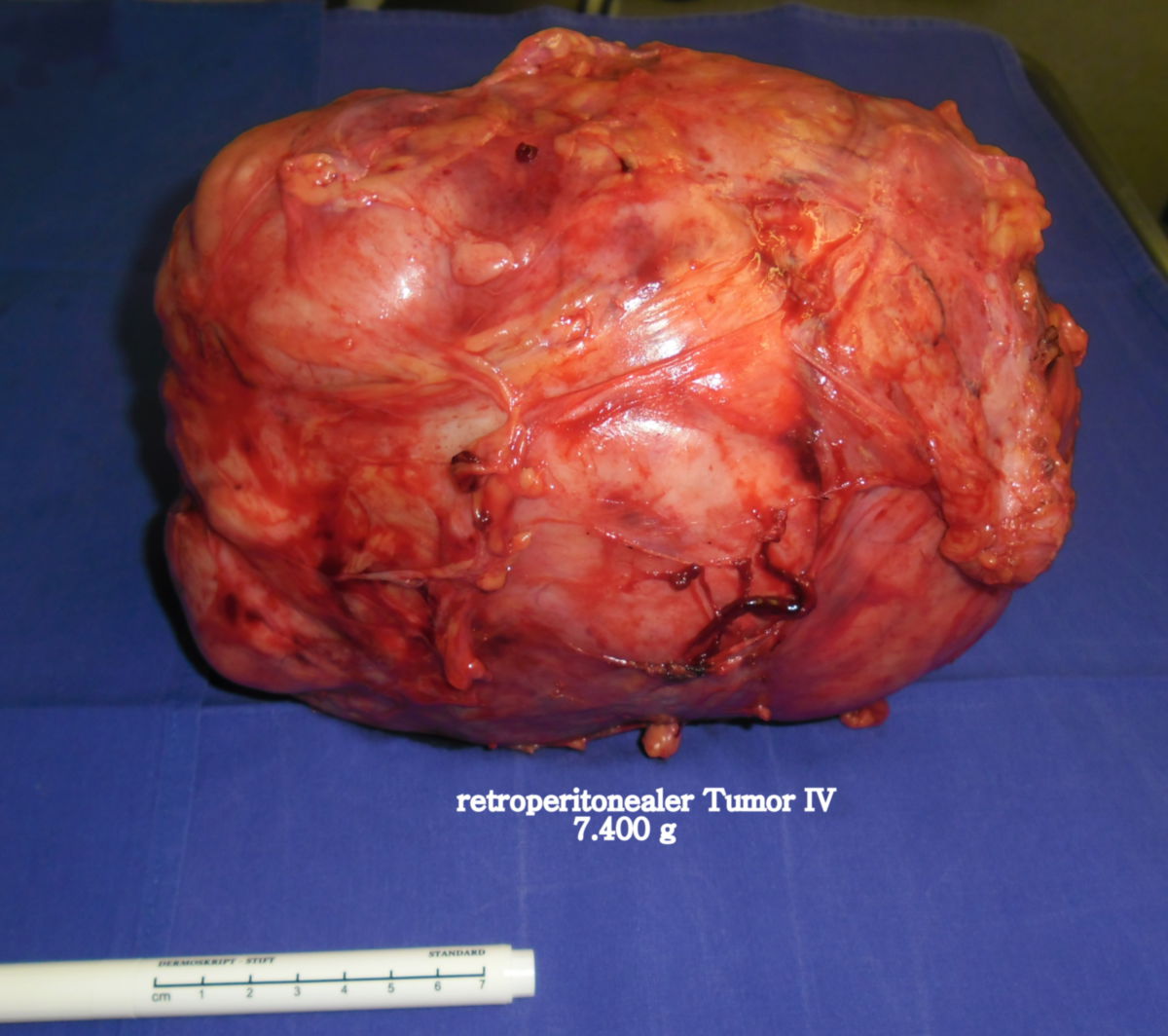
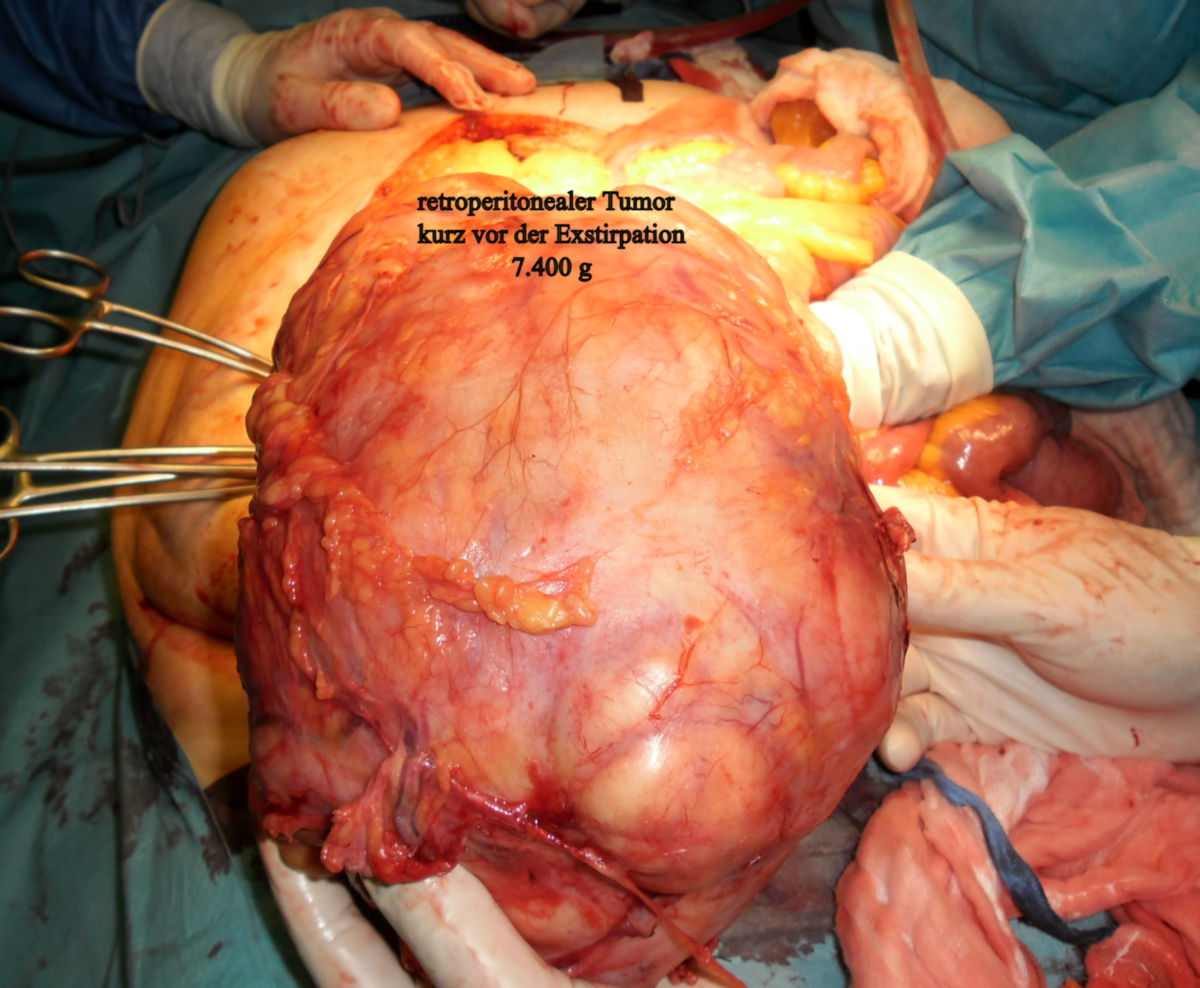
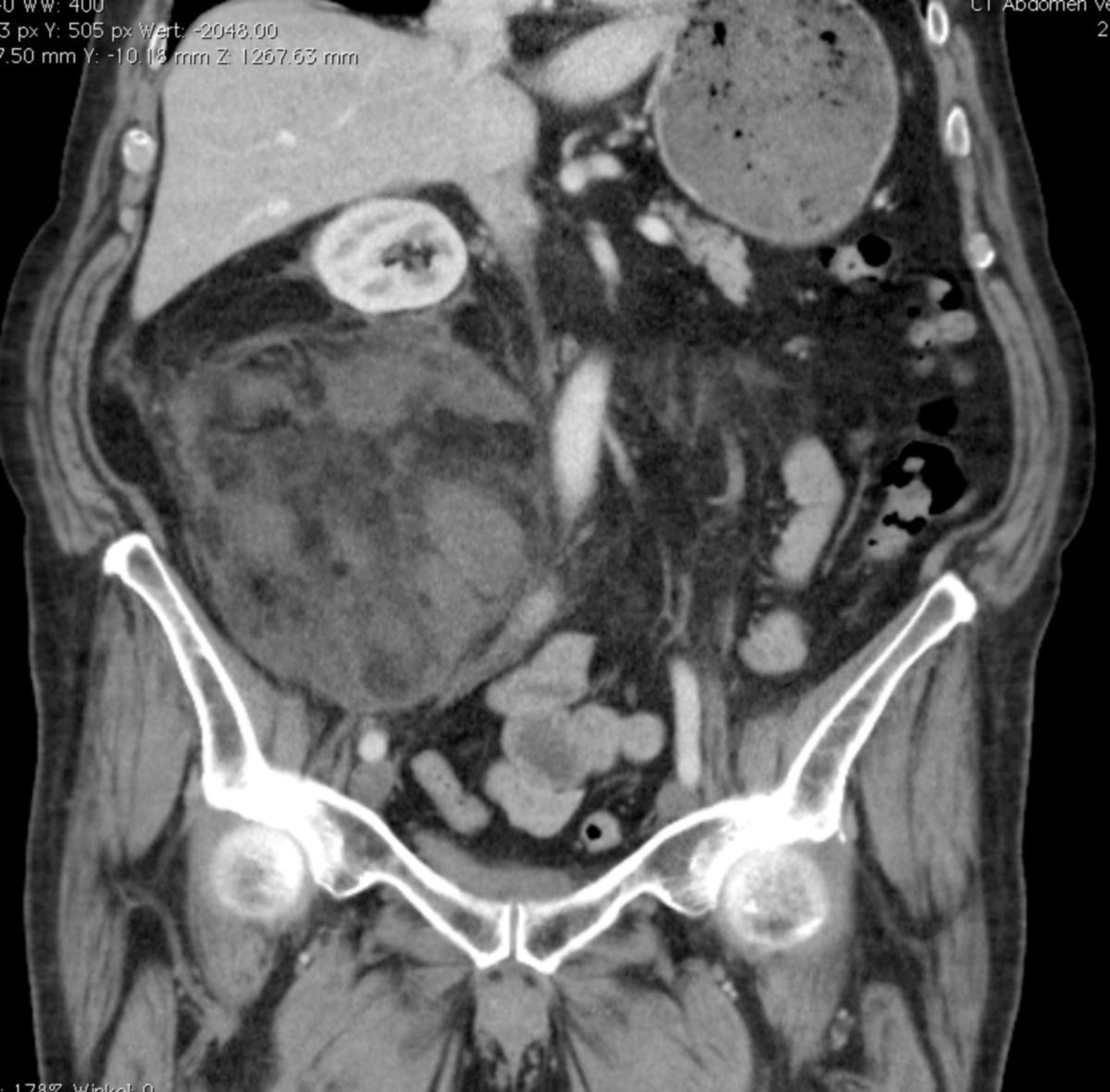
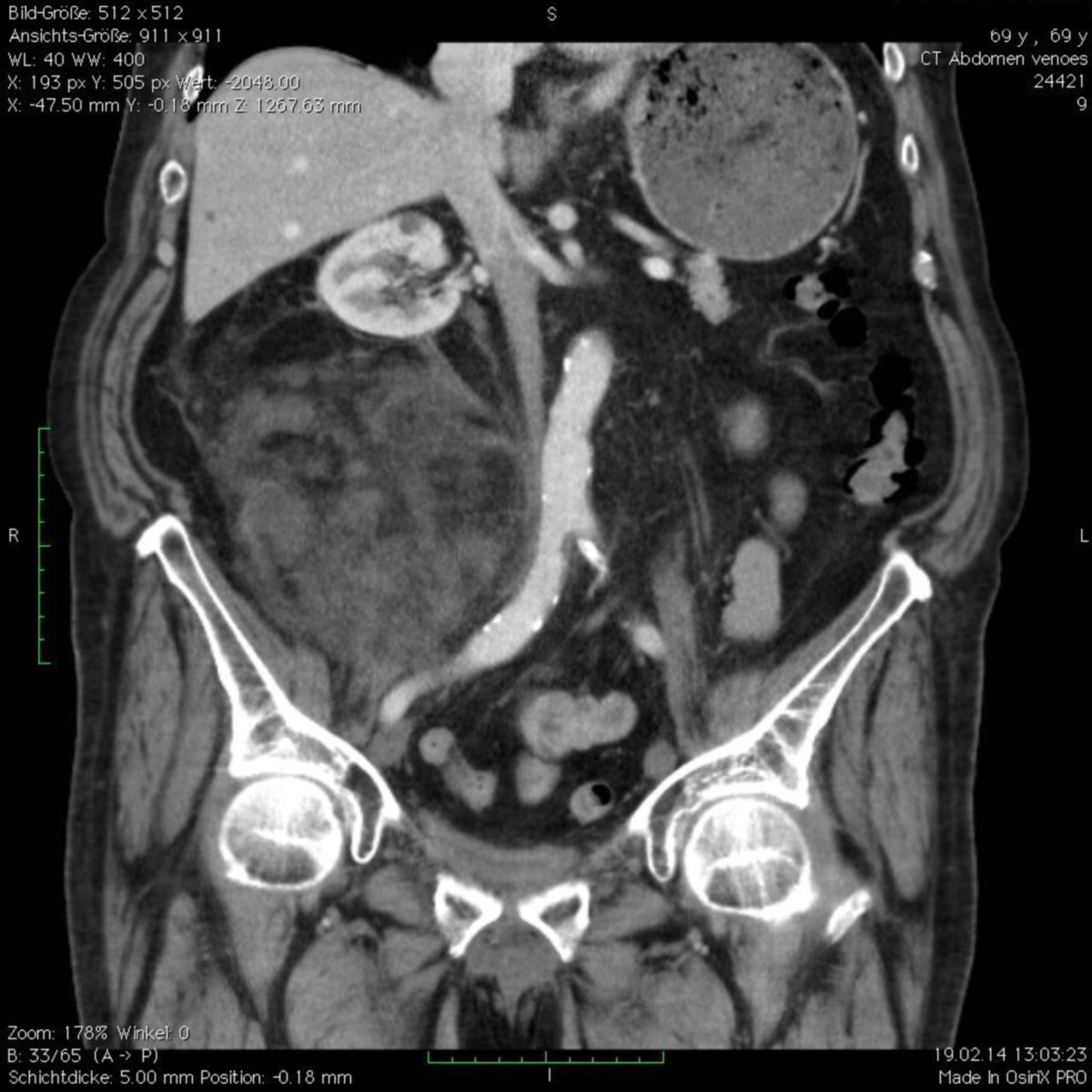
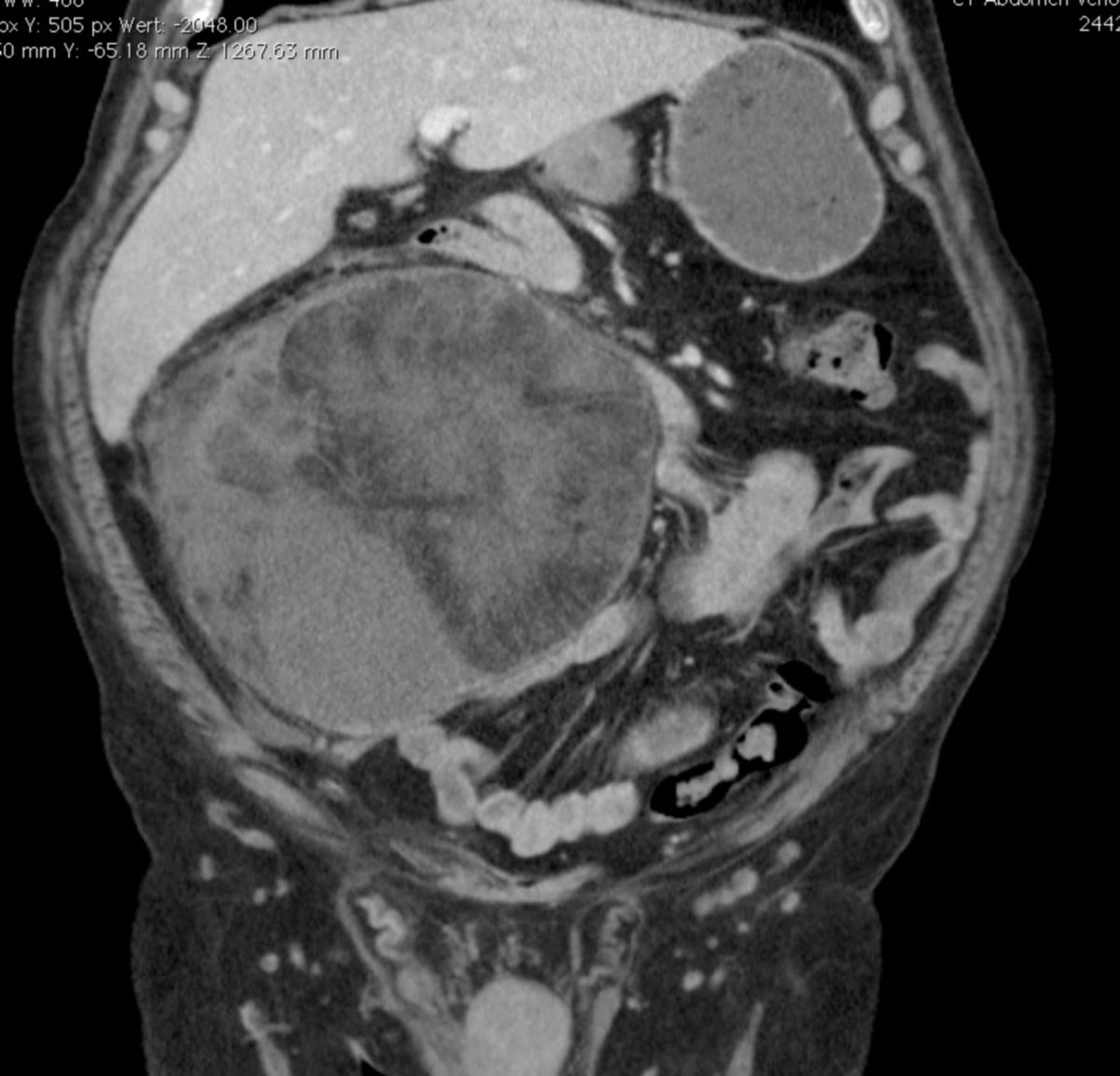
Liposarkome treten bevorzugt am Rumpf, den Extremitäten und im Retroperitoneum auf.
CT-Fallbeispiel
Makroskopie
Liposarkome zeichnen sich durch ihre gelbliche Farbe und ihre oft beträchtliche Größe aus. Myxoide Liposarkome zeigen typischerweise eine gelatinös-muköse Schnittfläche. Je nach Subtyp können zentrale Nekrosen, Hämorrhagien und Verkalkungen auftreten.
Klassifikation
Nach aktueller WHO-Klassifikation werden Liposarkome in folgende Haupttypen eingeteilt:
- atypischer lipomatöser Tumor/hoch differenziertes Liposarkom (ALT/WDLPS)
- dedifferenziertes Liposarkom (DDLPS)
- myxoides Liposarkom (MLPS)
- pleomorphes Liposarkom (PLPS)
Hoch differenziertes Liposarkom
Hoch differenzierte Liposarkome (auch atypischer lipomatöser Tumor, ALT) treten überwiegend im höheren Lebensalter auf. Histologisch kann aufgrund der hohen Differenzierung eine Verwechslung mit benignem Fettgewebe oder einem Lipom erfolgen. Charakteristisch sind atypische stromale Zellen mit vergrößerten, hyperchromen Kernen; molekularpathologisch finden sich häufig MDM2- und CDK4-Amplifikationen.
Der Tumor neigt zu Lokalrezidiven, metastasiert jedoch in der Regel nicht, sofern keine Dedifferenzierung vorliegt.
Dedifferenziertes Liposarkom
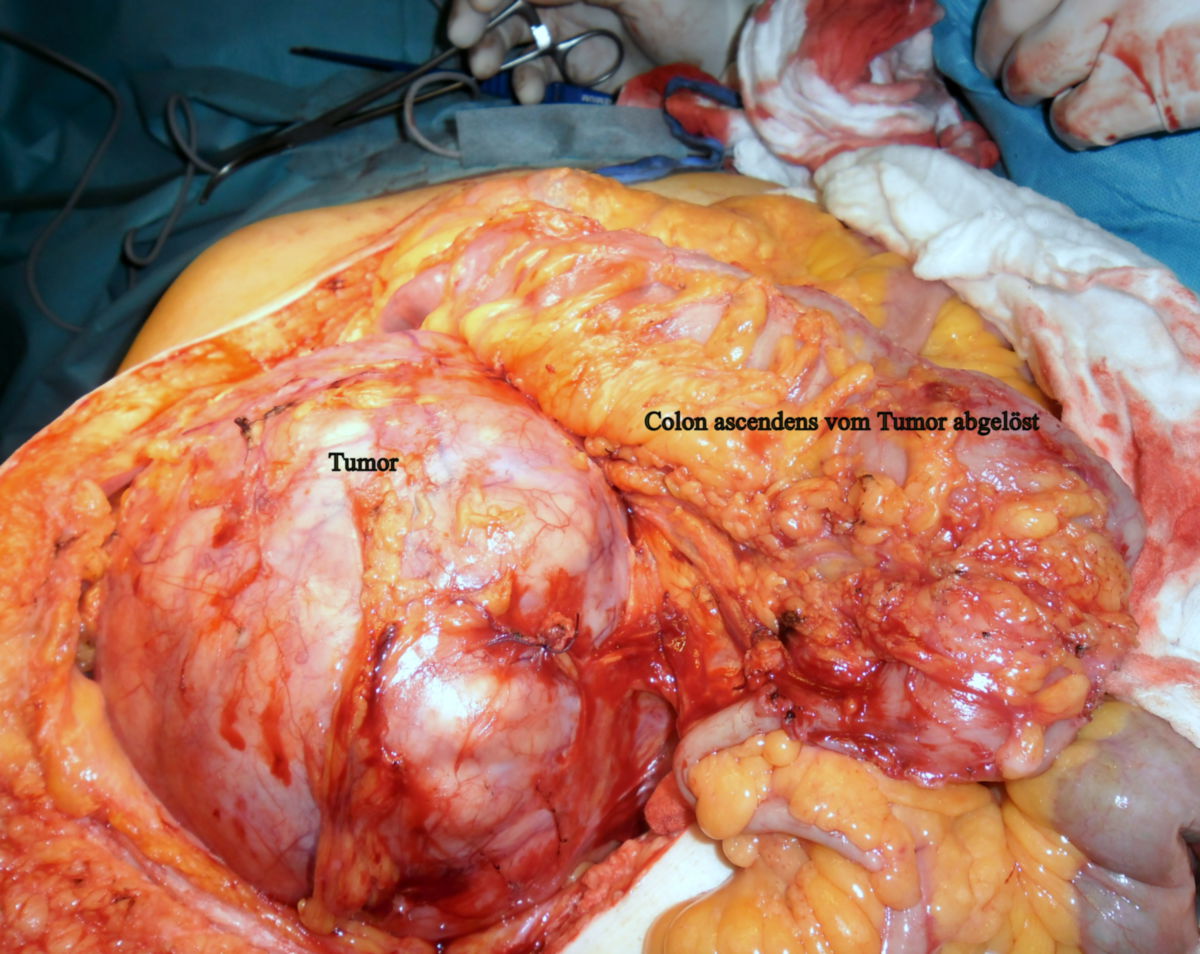
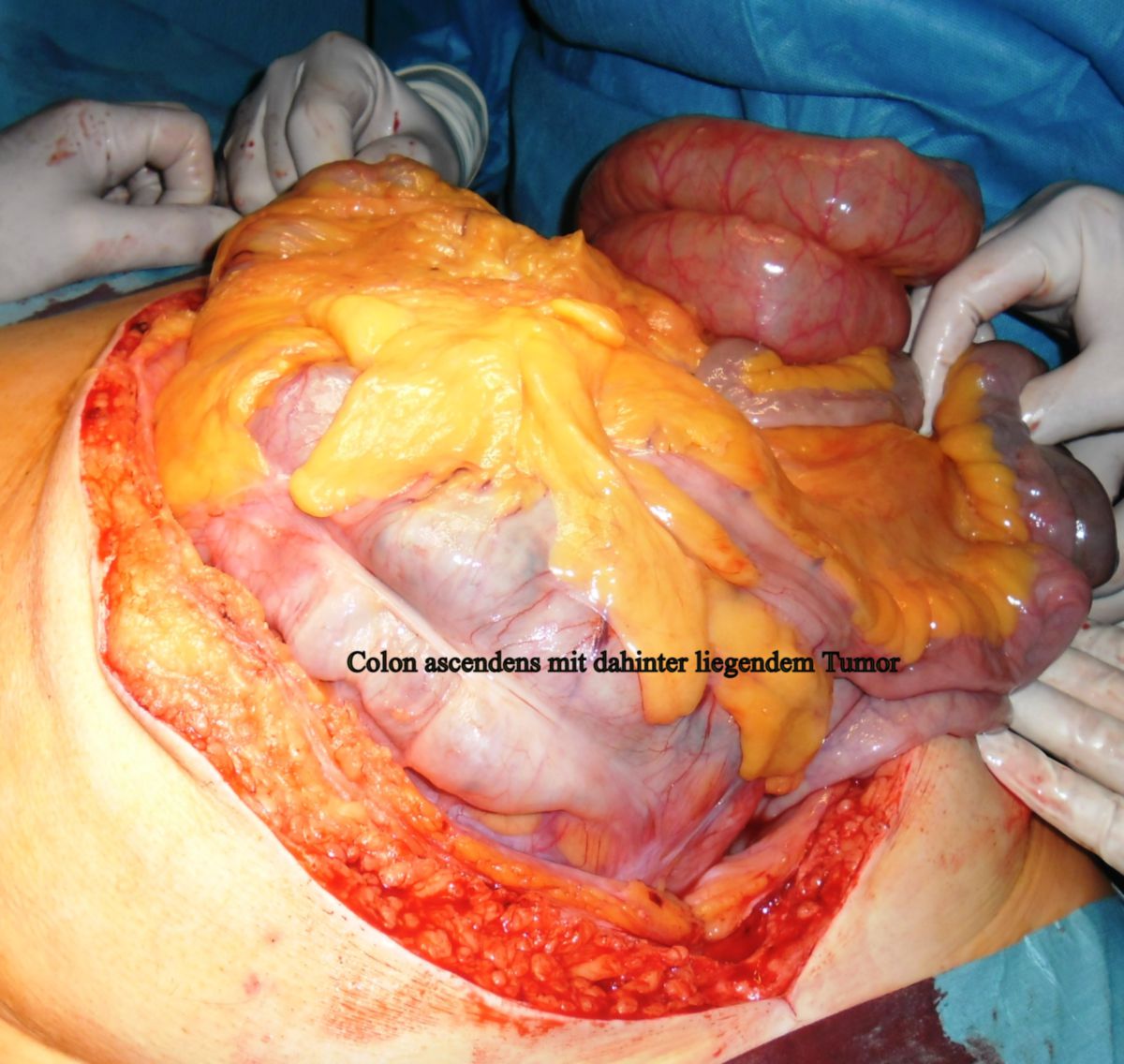
Das dedifferenzierte Liposarkom (DDLPS) entsteht meist auf dem Boden eines hoch differenzierten Liposarkoms und ist durch den Übergang in einen nichtlipogenen, meist hochgradigen Sarkomanteil gekennzeichnet. Es tritt bevorzugt im Retroperitoneum auf.
Im Vergleich zum hoch differenzierten Liposarkom weist das dedifferenzierte Liposarkom ein deutlich erhöhtes Risiko für Lokalrezidive und Metastasierung auf.
Myxoides Liposarkom
Das myxoide Liposarkom ist ein häufiger Subtyp und wird nach dem vorkommenden myxoiden (schleimigen) Stroma benannt, in dem fettvakuolenreiche Lipoblasten bzw. Prälipozyten zu finden sind. Die hühnerfußartig angeordneten Kapillaren stehen im engen Kontakt zu den Prälipozyten.
Diese Form des Liposarkoms ist geringgradig maligne und weisen eine vergleichsweise hohe Strahlensensibilität auf. Die Prognose ist mit einer 5-Jahres-Überlebensrate von etwa 70 % günstig.
Rundzelliges Liposarkom
Das rundzellige Liposarkom wird heute als hochgradige (rundzellige) Komponente des myxoiden Liposarkoms verstanden. Es ist durch gering differenzierte, runde Tumorzellen gekennzeichnet und mit einer erhöhten Metastasierungsrate assoziiert.
Pleomorphes Liposarkom
Das pleomorphe Liposarkom ist hoch maligne und hat mit einer 5-Jahres-Überlebensrate von etwa 20 % eine schlechte Prognose. Histologisch lassen sich bei dieser Form des Liposarkoms sehr variabel geformte Tumorzellen erkennen.
Therapie
Die Therapie des Liposarkoms besteht primär in der vollständigen chirurgischen Resektion mit dem Ziel einer R0-Resektion. Je nach Lokalisation und Subtyp kann eine adjuvante oder neoadjuvante Strahlentherapie sinnvoll sein, insbesondere bei myxoiden Liposarkomen. Eine Chemotherapie wird vor allem bei hochgradigen oder metastasierten Tumoren erwogen.
Als Whoops-Operation bezeichnet man die unbeabsichtigte marginale Entfernung eines vermeintlich benignen Weichteiltumors, der sich postoperativ als Sarkom herausstellt. In diesem Fall ist in der Regel eine Nachresektion in einem spezialisierten Zentrum erforderlich.
Bildquelle
- Bildquelle DICOM-Viewer: Roche, C., Bonaccio, E., & Filippini, J. (2016). The Cancer Genome Atlas Sarcoma Collection (TCGA-SARC) (Version 3) [TCGA-QQ-A5V2]. The Cancer Imaging Archive.