Autosomal-dominante polyzystische Nierenerkrankung







Englisch: autosomal dominant polycystic kidney disease, ADPKD
Definition
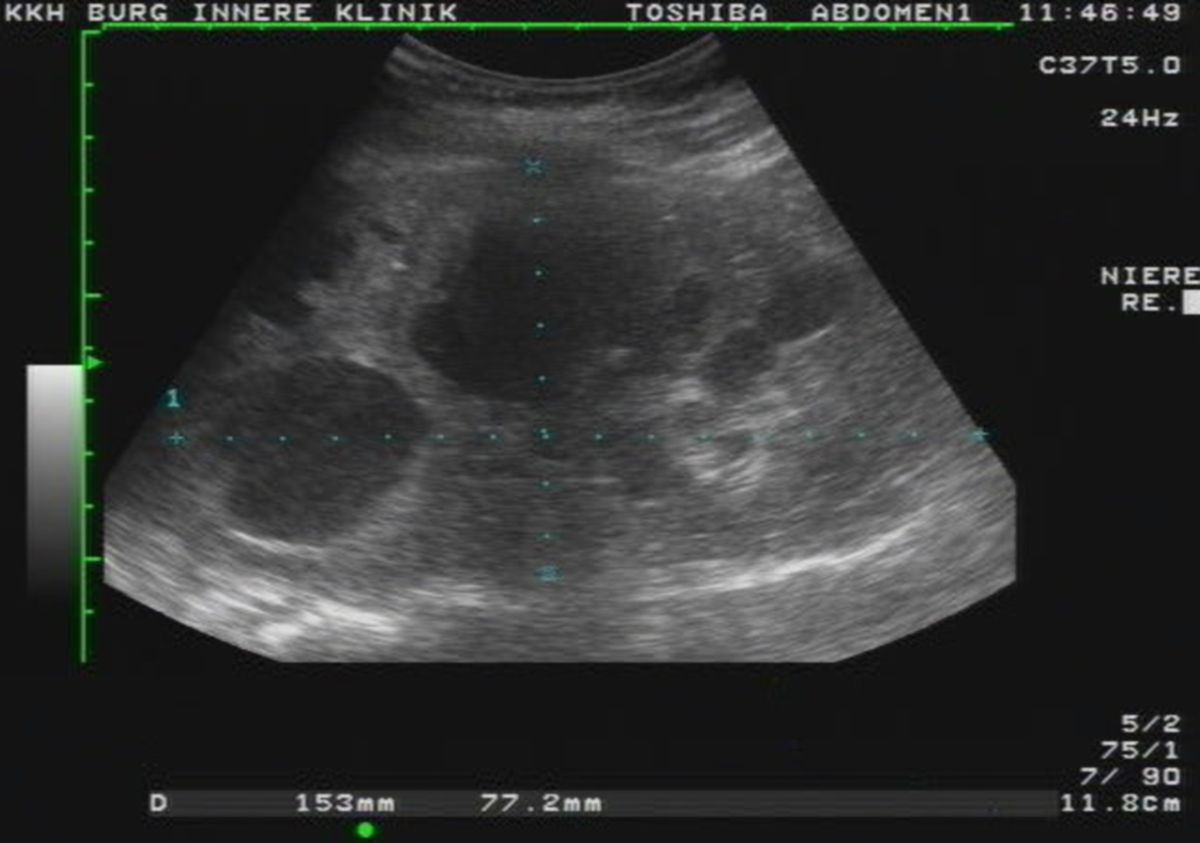
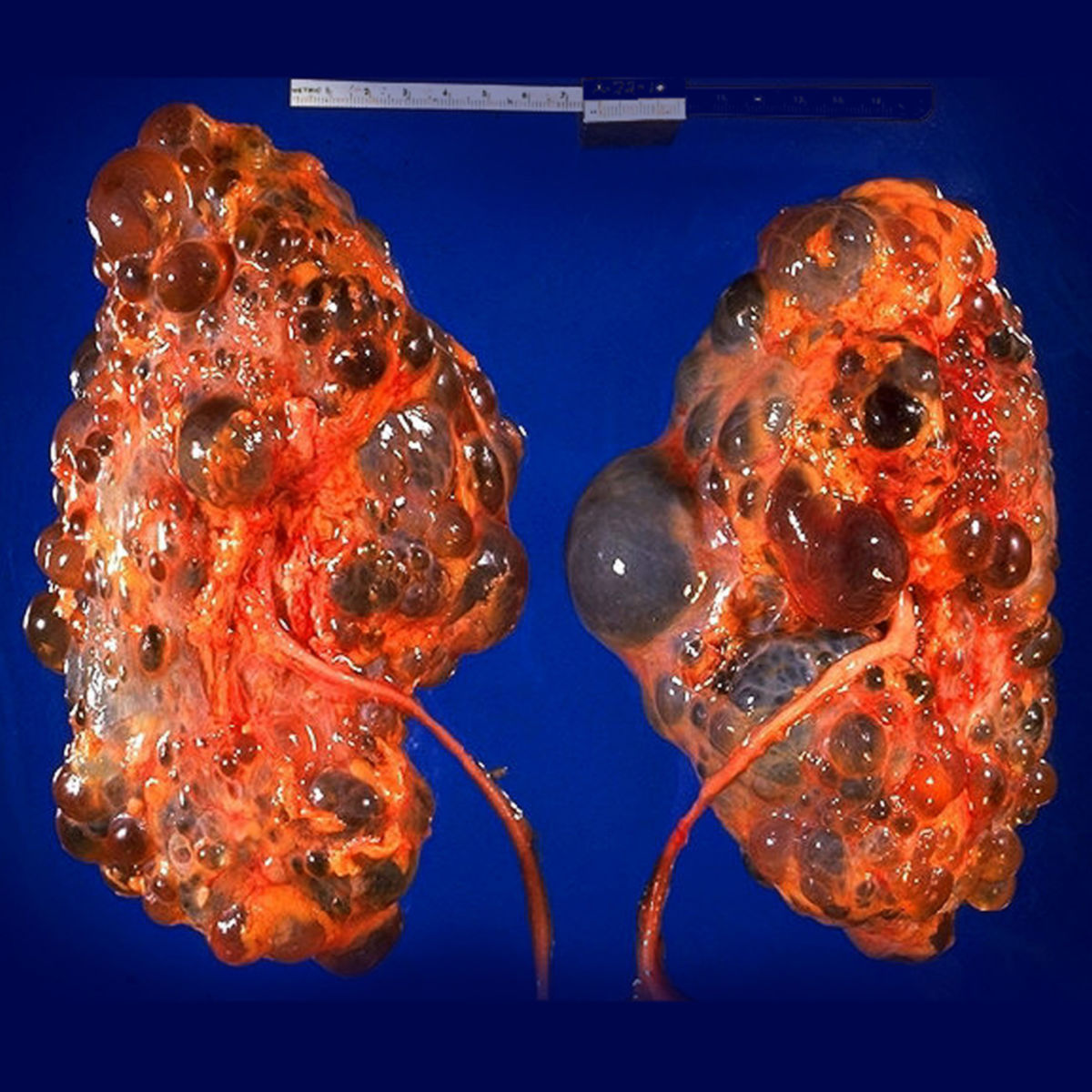
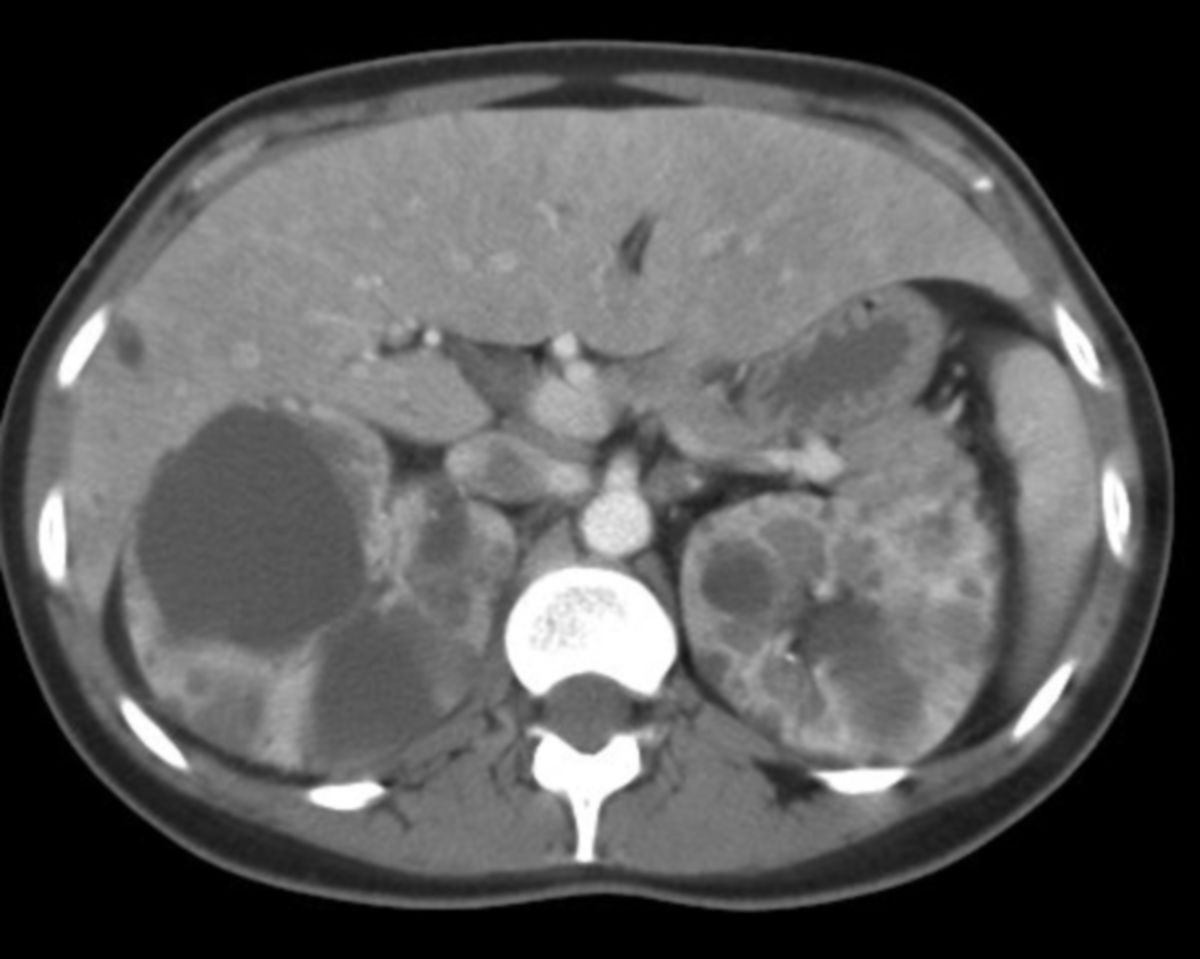
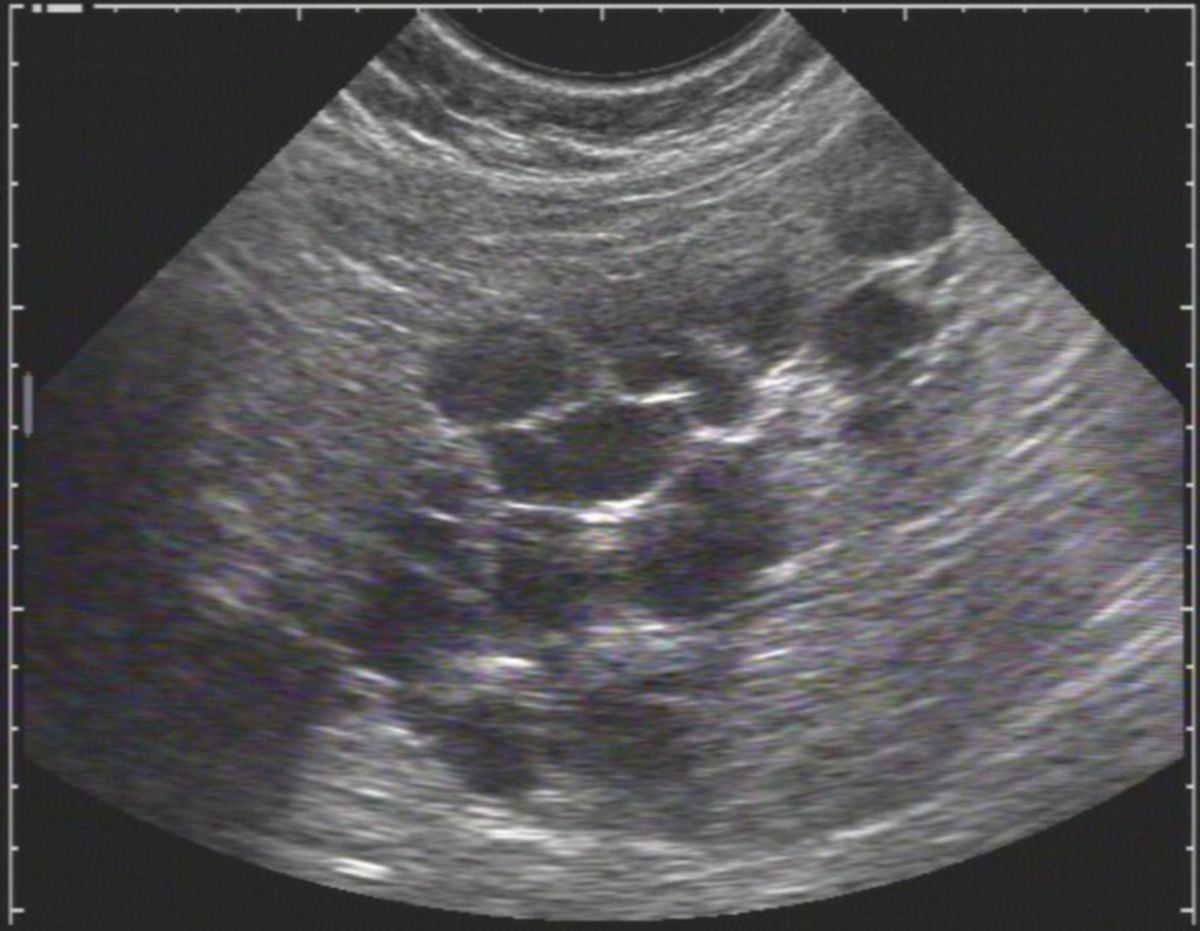
Die autosomal-dominante polyzystische Nierenerkrankung, kurz ADPKD, ist eine autosomal-dominant vererbte Erkrankung, die durch die Bildung von Zystennieren gekennzeichnet ist.
- ICD-10-Code: Q61.2
Ursache und Prävalenz
Es gibt verschiedene Mutationen, die eine ADPKD auslösen können. In 80 % der Fälle liegt auf Chromosom 16 ein defektes PKD1-Gen vor, das für Polycystin-1 kodiert. Das Gen PKD2, das für Polycystin-2 kodiert und sich auf dem Chromosom 4 befindet, zeigt bei 15 % der Betroffenen eine Mutation. Die Proteine Polycystin-1 und -2 spielen eine wichtige Rolle bei der Signalübertragung und der Ausbildung des Primärziliums der Tubulusepithelzellen. Dieses ermöglicht es den tubulären Zellen, die Flussraten zu messen. Mutationen können somit die Funktion der Zilien stören. Angenommen wird, dass die Proliferation und Differenzierung der Tubuluszellen mit der Flussrate zusammenhängen und dass eine Ziliendysfunktion eine zystische Veränderung auslösen kann.[1][2]
Bei den übrigen 5 % der Patienten ist eine genaue Genzuordnung nicht möglich. Bei der Prävalenz gibt es unterschiedliche Angaben, die zwischen 1 zu 300 und 1 zu 1.000 schwanken. Sicher ist, dass die Dunkelziffer höher ist und die ADPKD als häufigste lebensbedrohliche Erbkrankheit bezeichnet werden kann.
Symptome
Die Symptomatik ist bei allen Patienten nahezu gleich, lediglich in der Schwere der Symptome gibt es Unterschiede. Hier gilt die Regel, dass Patienten mit großen Nieren (>15 cm) häufiger und stärkere Beschwerden aufweisen. Langfristig kommt es zu einer Niereninsuffizienz und abschließend zu einem totalen Nierenversagen.
Hypertonie
In 60 bis 70 % entwickelt sich eine Hypertonie. Sie wird vermutlich dadurch ausgelöst, dass Zysten ein Nierengefäß komprimieren, so dass im distalen Abschnitt des Gefäßes der Blutdruck sinkt. Dieser Blutdruckabfall wird von der betroffenen Niere registriert, was eine Ausschüttung von Renin und damit eine Steigerung des Blutdrucks zur Folge hat.
Chronischer Schmerz
Da die Zysten auch die umgebenden Nerven komprimieren können, kann es zu chronischen Schmerzen kommen. Charakteristisch strahlen diese in den Rücken, die Flanken und die Leisten aus.
Hämaturie
Bei jedem zweiten Patienten mit ADPKD kann Blut im Urin nachgewiesen werden (Hämaturie). Die Menge kann jedoch variieren, so dass es sich in manchen Fällen nur um mikroskopische Mengen handelt (Mikrohämaturie).
Harnwegsinfektion
Bei manchen Patienten kommt es vermehrt zu Harnwegsinfekten, wobei hier Frauen typischerweise häufiger betroffen sind. Das erhöhte Risiko lässt sich so erklären, dass die Zysten einen guten Sammelpunkt für Bakterien darstellen.
Nierensteine
20 bis 30 % der ADPKD-Patienten haben ein Nierensteinleiden. Dies liegt unter anderem daran, dass die Zysten den Abfluss des Urins behindern und sich daher schneller Kristalle bilden können.
Extrarenale Manifestationen
Auch andere Organe können von der ADPKD betroffen sein. So können Zysten in der Leber, seltener auch in der Milz und im Pankreas auftreten. Durch Strukturschwächen zerebraler Arterien ist das Auftreten von Aneurysmen möglich. Auch Colondivertikel, Leistenhernien und Aorteninsuffizienzen kommen bei ADPKD-Patienten häufiger vor.
Diagnostik
Die Diagnose erfolgt per Bildgebung. Die Progressionsrate der Niereninsuffizienz kann mithilfe der Mayo Imaging Classification abgeschätzt werden.
Therapie
Die Behandlung der ADPKD ist rein symptomatisch, da der zugrundeliegende Gendefekt zur Zeit (2025) nicht beseitigt werden kann. Die Krankheitsprogression lässt sich durch den Vasopressin-Antagonisten Tolvaptan in individuell unterschiedlichem Maß verzögern. Im Rahmen dieser Therapie ist eine regelmäßige Überprüfung der Leberfunktion notwendig. Darüber hinaus werden Antihypertensiva und Antibiotika gegeben, falls eine Hypertonie bzw. ein Harnwegsinfekt vorliegt.
Im fortgeschrittenen Stadium ist eine Dialyse indiziert. Eine Heilung kann nur durch eine Nierentransplantation erreicht werden.
Quellen
- ↑ Urologielehrbuch.de; Autosomal-dominante polyzystische Nierenerkrankung (ADPKD); abgerufen am 08.05.2023
- ↑ MSD Manual; Autosomal-dominante polyzystische Nierenerkrankung (ADPKD); abgerufen am 08.05.2023