HMG-CoA-Lyase




Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenSynonyme: β-Hydroxy-β-Methylglutaryl-Coenzym-A-Lyase, Hydroxymethylglutaryl-CoA-Lyase, 3-Hydroxy-3-Methylglutaryl-CoA-Lyase
Definition
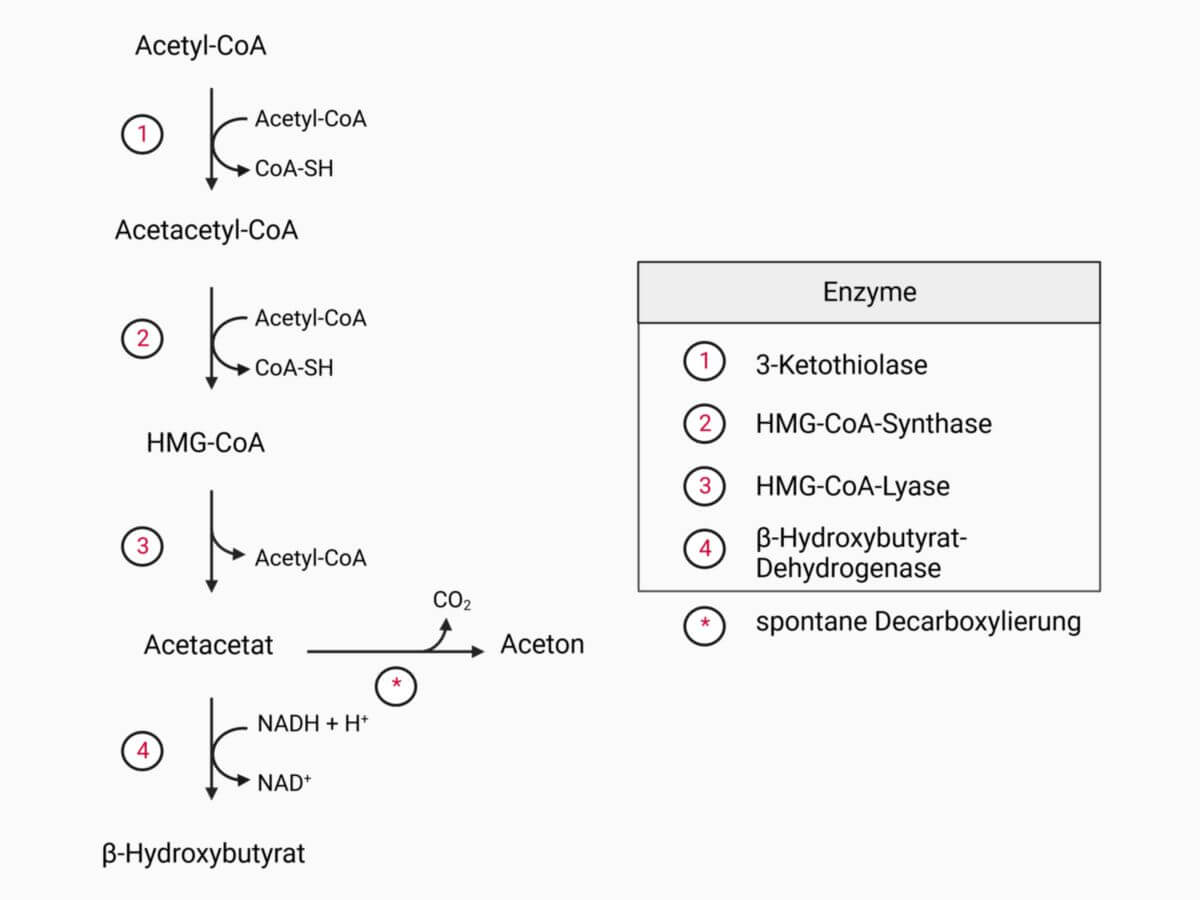
Die HMG-CoA-Lyase ist ein Enzym der EC-Klasse 2.4 (Lyasen) und Teil der Ketonkörpersynthese. Es katalysiert die Spaltung von HMG-CoA in Acetyl-CoA und den Ketonkörper Acetoacetat.
Hintergrund
Um im Falle eines Glucosemangels die zelluläre Energieversorgung aufrechtzuerhalten, können in den Mitochondrien der Leber aus abgebauten Lipiden Ketonkörper synthetisiert werden. Die Leber kann Ketonkörper produzieren, jedoch nicht selbst verwerten, sodass sie ins Blut abgegeben und zu den Endorganen transportiert werden.
Genetik
Das Protein wird vom HMGCL-Gen auf Chromosom 1 am Genlokus 1p36.11 kodiert und hat eine Länge von 235 Aminosäuren.
Biochemie
Im letzten Schritt der Lipolyse (Abbau von Fettsäuren) entsteht Acetyl-CoA. Für die Ketonkörpersynthese reagieren mithilfe der 3-Keto-Thiolase zwei Acetyl-CoA-Moleküle zu Acetacetyl-CoA. Unter Katalyse durch die HMG-CoA-Synthase wird ein weiteres Acetyl-CoA hinzugefügt, wodurch β-Hydroxy-β-methylglutaryl-CoA (HMG-CoA) entsteht. Als letzter Schritt der Ketogenese wird nun HMG-CoA durch die HMG-CoA-Lyase in Acetyl-CoA und den Ketonkörper Acetoacetat gespalten. Acetoacetat wird anschließend meist durch die β-Hydroxybutyrat-Dehydrogenase zu dem im Blut am häufigsten vorkommenden Ketonkörper β-Hydroxybutyrat reduziert.
Klinik
In sehr seltenen Fällen kommt es zu einem HMG-CoA-Lyase-Mangel. Es handelt sich um eine autosomal-rezessiv vererbte Erkrankung. In Fastenperioden oder bei erhöhtem Energiebedarf (z.B. bei Infektionen) können sich Hypoglykämien mit Ketonkörpermangel und Azidose entwickeln.
Literatur
- Löffler, Petrides, Biochemie und Pathobiochemie, 9. Auflage, Springer Verlag
- orpha.net – 3-Hydroxy-3-Methylglutarazidurie, abgerufen am 19.09.2023
- GeneCards – 3-Hydroxy-3-Methylglutaryl-CoA Lyase, abgerufen am 19.09.2023