Atypischer teratoider/rhabdoider Tumor





Trainier deine Lernmuskeln!
Mit Flash Cards, Quiz und mehr
LoslegenAbkürzung: ATRT, AT/RT
Englisch: atypical teratoid rhabdoid tumor
Definition
Beim atypischen teratoiden/rhabdoiden Tumor, kurz ATRT handelt es sich um eine embryonale Raumforderung des Gehirns, die vorwiegend im Kleinkindesalter auftritt. Der ATRT zeichnet sich durch eine hochgradige Malignität aus.
Verbreitung
Mit etwa 2 bis 3 % aller frühkindlichen Hirntumoren ist der ATRT relativ selten. Er tritt in den meisten Fällen sporadisch auf. Kommt er familiär gehäuft vor, liegt oft ein Rhabdoid-Prädispositions-Syndroms vor.
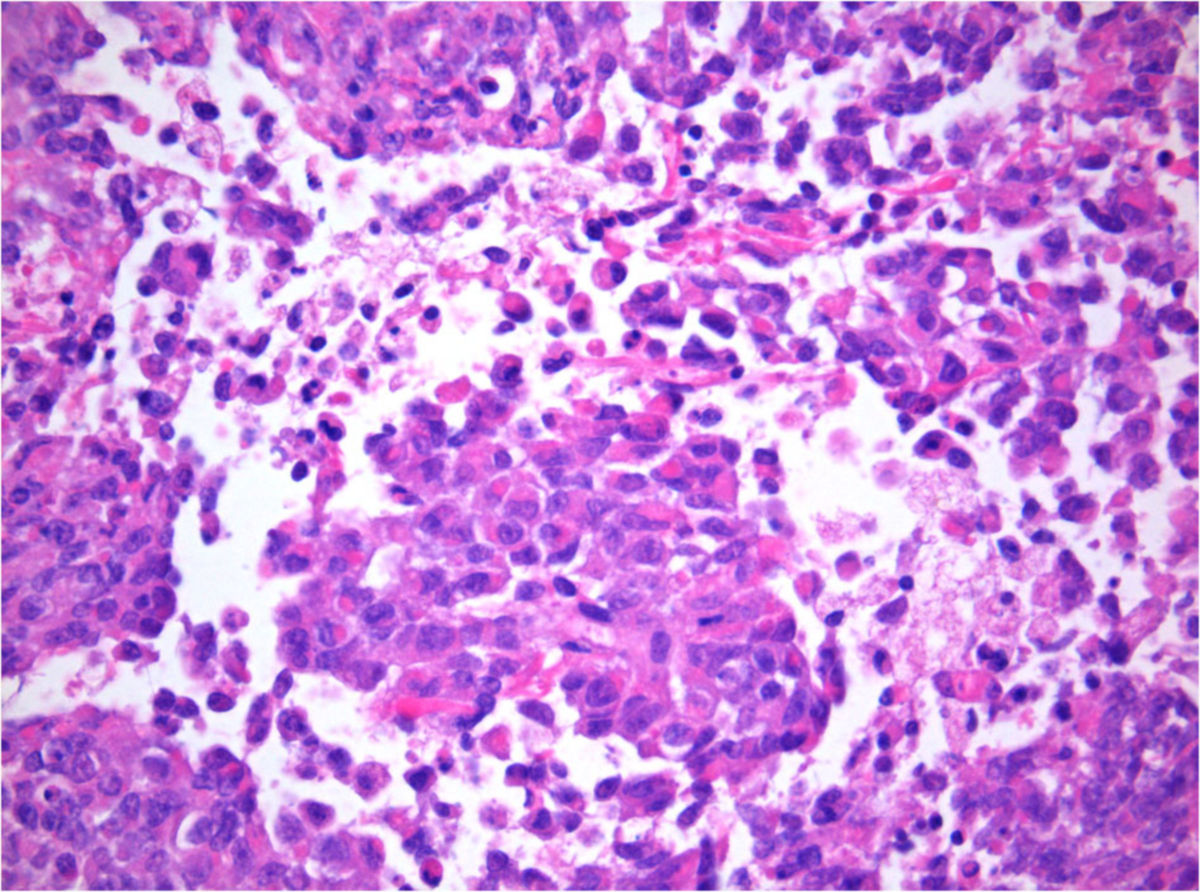
Histologie
- rhabdoide Tumorzellen: große Zellen mit am Rand liegenden Nukleolen
- sehr hohe Mitoserate
- infiltratives Wachstum
- histologische Ähnlichkeit mit dem Rhabdomyosarkom
Genetik
- Verlust des langen Arms von Chromosom 22 mit konsekutivem Verlust des hSNF5/INI-1 Gens[1]
- Keimbahnmutation des SMARCB1-Gens finden sich bei rund 40 % der Betroffenen
Symptome
Die Symptomatik des ATRT ist stark abhängig von der genauen Lage im Gehirn. Praktisch immer leiden Patienten unter starker Übelkeit, Schwindel, Erbrechen und Antriebslosigkeit.
Diagnose
- Computertomografie (CT)
- Magnetresonanztomografie (MRT)
- Entnahme einer Biopsie (erst diese erlaubt eine pathologisch eindeutige Diagnose)
Therapie
Aufgrund der oft sehr ungünstigen Lage im ZNS ist eine vollständige operative Resektion häufig nicht möglich. Es steht des Weiteren eine Chemotherapie zur Verfügung. Eine Bestrahlung kann erst ab dem 3. Lebensjahr durchgeführt werden.
Prognose
Der ATRT hat eine ungünstige Prognose. Selbst nach vollständiger Resektion des ATRT kommt es häufig zu Rezidiven. Eine adäquate Einschätzung der Prognose ist wegen der Seltenheit des Tumors und des damit verbundenen Datenmangels schwierig. Die Prognose hängt u.a. von folgenden Faktoren ab:
- Lebensalter (bessere Prognose bei Alter > 3 Jahre)[2][3]
- Metastasierung und Tumorwachstum[3]
- Diagnosezeitpunkt
- Anatomische Lage[4]
- Behandlungsregime[1][2]
Die 5-Jahres-Überlebensrate variiert in verschiedenen Publikationen, beträgt aber etwa 40%.[4][5] Die Strahlentherapie scheint die wichtigste Komponente des multimodalen Therapiekonzeptes darzustellen. Sie ist jedoch vor allem bei Kindern unter 3 Jahren keine Option.[1]
Quellen
- ↑ 1,0 1,1 1,2 [1] Ginn KF, Gajjar A. Atypical teratoid rhabdoid tumor: current therapy and future directions. Front Oncol. 2012;2:114.
- ↑ 2,0 2,1 [2]Tekautz TM, Fuller CE, Blaney S, et al. Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J Clin Oncol 2005;23(7):1491-9.
- ↑ 3,0 3,1 [3]Dufour C, Beaugrand A, Le Deley MC, et al.: Clinicopathologic prognostic factors in childhood atypical teratoid and rhabdoid tumor of the central nervous system: a multicenter study. Cancer 118 (15): 3812-21, 2012.
- ↑ 4,0 4,1 [4]Chi SN, Zimmerman MA, Yao X, et al. Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor; J Clin Oncol 2009;27:385–9.
- ↑ [5] Woehrer A, Slavc I, Waldhoer T, Heinzl H, et al. Incidence of atypical teratoid/rhabdoid tumors in children: a population-based study by the Austrian Brain Tumor Registry, 1996-2006; Cancer. 2010; 116(24):5725-32.